第三代和第四代氟喹诺酮的分析方法
2019-10-12张姝李红冯连顺编写刘明亮郭慧元审校
张姝, 李红, 冯连顺 编写 刘明亮, 郭慧元 审校
(1 武汉谱尼科技有限公司,武汉 430014;2 中国医学科学院北京协和医学院医药生物技术研究所 100050)
1 前言
自第一个喹诺酮——萘啶酸于1963年被美国食品药品监督管理局(FDA)批准用于治疗细菌感染,尤其是上世纪80年代初Koga等结合吡哌酸和氟甲喹的结构特征成功开发出喹诺酮发展史上具有划时代意义的6-氟-7-哌嗪基取代的首个氟喹诺酮——诺氟沙星以来,这类药物的发展日新月异,抗菌谱和药动学性质等得到显著改善,更具特点的新品种不断问世。氟喹诺酮可通过与细菌DNA促旋酶或拓扑异构酶IV催化结构域中的喹诺酮耐药决定区(QRDR)键来干扰DNA的正常复制与转录,启动细胞死亡机制导致细胞凋亡。目前,氟喹诺酮已发展成为仅次于头孢菌素的第二大类抗感染化疗药物,对革兰氏阳性菌、阴性菌、非典型性细菌、支原体、衣原体和军团杆菌等具有广谱抗菌活性,为人类的生命健康做出了巨大贡献。目前,喹诺酮已发展到第四代,通常而言新一代喹诺酮比前一代抗菌谱更广、体内活性更高、药代动力学性质更优和毒副作用更低。
研究表明,氟喹诺酮的活性与其浓度息息相关,其药代动力学性质可能受患者尤其是危重病人、肾功能不全患者和住院患者自身影响。其中,药时曲线下面积(AUC)与MIC比值和血药峰浓度(Cmax)与MIC比值为描述氟喹诺酮活性的最重要药代动力学参数,对二者的优化可给予患者有效药物治疗,进而降低耐药性发生的几率、提高治愈率。
高效液相色谱(HPLC)与各种检测器联合使用是研究氟喹诺酮药代动力学性质的常用方法,其中HPLC与紫外(HPLC-UV)、与荧光检测器(HPLCFLD)和与质谱(HPLC-MS)联用最为常见。值得一提的是,HPLC-MS联用技术使微量检测成为可能。毛细管电泳法(CE)与UV或FLD联合使用是检测氟喹诺酮的另一重要分析方法。而药物制剂中的药物水平可用HPLC或其它检测手段如紫外光谱、线性扫描伏安法甚至核磁共振(NMR)检测。样品在检测前往往需要进行适当的预处理,如蛋白沉淀、萃取、溶解、过滤和稀释等。药物制剂的预处理最为简单仅需要稀释即可,而生理流体如血液、胆汁、唾液和尿液及组织匀浆的处理则相对复杂,这主要是由于内源性物质可能会出现在色谱图或电泳图上,干扰分析结果。因此,选择合理的样品处理方法和分离条件(合适的溶剂或缓冲液)对结果的可靠性而言至关重要。本文将着重介绍近年来所发展的第三代和第四代喹诺酮的分析方法及样品的预处理方法,为分析人员从事相关研究提供帮助。
2 第三代氟喹诺酮
左氧氟沙星(LVFX)、巴洛沙星(BLFX)、帕珠沙星(PZFX)和司帕沙星(SPFX)是第三代氟喹诺酮的杰出代表。LVFX不仅可用于治疗社区获得性肺炎(CAP)、急性上颌窦炎和慢性支气管炎急性发作,还可用于消除标准疗法无法消除的幽门螺旋杆菌。LVFX的生物利用度完全,故口服和静脉注射等效。LVFX的代谢较差,在给药48h后约87%的LVFX经尿液清除。LVFX主要的代谢产物为N-氧化物和去甲基LVFX,但二者均没有活性。BLFX对革兰氏阳性菌如耐多药葡萄球菌和肺炎球菌具有良好的抗菌活性,其在肾中的代谢产物为葡萄糖醛酸和去甲基BLFX衍生物。PZFX对革兰氏阴性菌具有优秀的活性,对肝组织、胆囊组织和胆汁具有良好的渗透性,提示本品对肝病患者而言可能大有作为。SPFX的体外抗分枝杆菌和革兰氏阳性菌如肺炎链球菌和其它链球菌及葡萄球菌的活性优于环丙沙星(CPFX)。
2.1 左氧氟沙星
LVFX的定量分析方法众多(表1),且大多数基于HPLC,这是由于LVFX在水中具有良好的溶解度。最常用的检测器有UV和FLD,而含量较低时可用MS检测。在LVFX的MS/MS分析中,经常采用m/z 362.7→261.2、m/z 362.1→318.1和m/z 362.2→261.2的质谱多反应监测(MRM)模式。在单MS分析中,经常可观测到m/z 362→318。除MS外,还可采用光电二极管阵列检测器(PDA)进行检测。流动相通常为水或缓冲溶液和有机溶剂,并加入三乙胺(TEA)以改善峰形、消除拖尾。TEA的含量通常低于<1%,且流动相的pH需显弱酸性(可用磷酸调pH)。LVFX含有带负电荷的羧基,故离子对试剂如TEA的加入可显著地提高分离度。其它极性组分可为磷酸盐(钠盐或钾盐)缓冲溶液,浓度在10~30 mmol/L。最常用的有机相为乙腈(ACN),等度洗脱时其浓度通常为14~43%,梯度洗脱时其浓度随时间变化而变化。在亲水相互作用液相色谱法(HILIC)中,ACN的浓度可达80%以上。
在反相HPLC(RP-HPLC)中,主导型的柱子为C18柱,但有时也使用C8和C4柱。Watabe等评价了各种类型的柱子如C18和C8柱在分析LVFX和PZFX时的效果,发现C8柱的效果优于C18柱,这可能是由于C8柱的空阻低于C18柱所致。LVFX和PZFX均含有7-氧代-吡啶并[1,2,3-de]-1,4-苯并嗪-6-羧酸结构单元,区别在于LVFX的C-10位为4-甲基哌嗪基,而PZFX的C-10位为1-氨基环丙基。这些基团的存在可能使二者与固定相表面的结合作用更强,分离效果更高。Fang等采用C4柱对LVFX、利福平和异烟肼等进行了分离,发现与C18柱相比,C4柱对这些极性迥异的物质分离效果更高。丁基键合固定相可缩短非极性物质的分析时间,但对极性物质影响不大,且与长链键合固定相相比,丁基键合固定相的分离度得以保持。亲水作用色谱(HILIC)柱也可用于LVFX的分析,其最大的优点是能够有效分离反相作用色谱中难以保留的离子型化合物。HILIC可用常见的反相流动相,但有机溶剂含量较高,故更适用于MS检测。除ACN外,甲醇也是常见的有机相,而且可用于等度洗脱和梯度洗脱。硫酸铜(CuSO4)和L-亮氨酸,及甲酸(用于MS检测)、十二烷基硫酸钠(SDS)、四丁基醋酸(TBAA)、柠檬酸、醋酸铵、四正丁基溴化铵(TBAmBr)和L-异亮氨酸组成的手性流动相添加剂(CMPA)也可作为流动相组分使用。Liang等研究结果表明,流动相中的SDS可延长LVFX、加替沙星(GTFX)、莫西沙星(MXFX)和曲伐沙星(TVFX)的保留时间,而25 mmol/L的磷酸缓冲液和离子对试剂(TBAA 10 mmol/L)可改善峰型,这可归因于这种流动相有可能能够阻止固定相上的硅醇基与喹诺酮的氨基发生二次交互作用。为获取立体选择性,可直接向流动相中加入手性配体交换试剂,如CuSO4和L-亮氨酸或L-异亮氨酸的加入可立体专一性测定LVFX。二价铜离子(Cu2+)、L-亮氨酸和水可形成复合物,该复合物可与LVFX及其R-对映异构体结合。这些螯合物具有不同的构型,保留时间不尽相同,可用于检测药物剂型中的光学纯度。对多种氨基酸如L-亮氨酸、L-苯丙氨酸、L-丝氨酸和L-丙氨酸等的评价结果显示,L-亮氨酸的分离度最高。Devi等报道了氧化降解的杂质分析方法,但非立体专一性,故其应用受到了一定的限制。
毛细管电泳法(CE)也可用于LVFX的检测,该法不仅用量少,而且适用范围广,可用于人尿液、片剂或水中LVFX的检测。该法既可用于溶液,也可用于非溶液。对溶液而言,pH在8左右时最优,pH的改变将影响检测器的相应,且pH<2.5时溶液可能会与毛细管壁发生相互作用。与柱色谱分离法相比,CE分离法更为复杂,其分离度受多种因素如pH、电压、温度和毛细管长度等影响,而且样品中的杂质可能会被毛细管壁吸收导致分离时间延长。
紫外-可见光吸收光谱法也被于LVFX的检测,但该法仅被用于纯药物或药物制剂的分析。LEVX可直接用于检测,也可与溴酚蓝(BPB)或溴甲酚绿(BCG)络合后在用于检测。光谱法在检测市售制剂、人尿和血清时,需应用荧光检测器,且SDS胶束具有加强荧光的效果。除紫外-可见光吸收光谱法外,NMR、方波阳极溶出伏安法和同步扫描固体基质室温磷光法也可用于LVFX的检测,但这些方法极少使用,且仅用于制剂的检测。
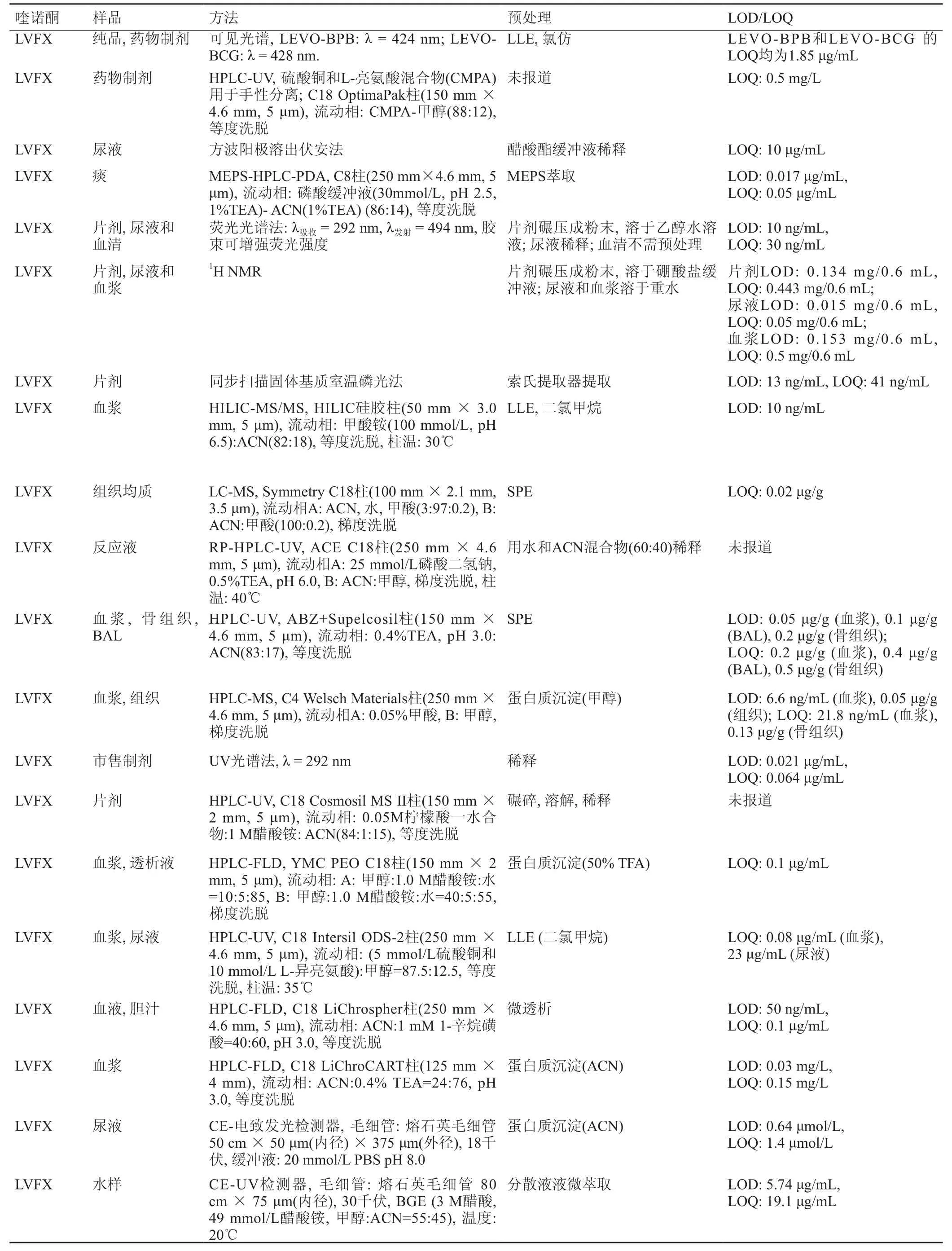
表1 LVFX的检测方法

续表 表1 LVFX的检测方法
上述的所有检测方法均需要对样品进行预处理,样品包括血清、血浆、血液、尿液、痰、组织、支气管肺泡灌洗(BAL)、胆汁、水、渗析液、反应液和药物制剂等。蛋白质沉淀用到的主要溶剂有ACN、甲醇、三氟乙酸(TFA)或ACN与甲醇的混合液或高氯酸与甲醇的混合液等。Watabe等发现采用ACN或乙醇做蛋白质沉淀时会导致峰很宽或很小;用甲醇时,上清液不够清澈;用6%的高氯酸溶液,则回收率低;用6%的高氯酸和甲醇的混合液,则可解决回收率的问题,这可能由于甲醇的加入使得药物与蛋白质共同被萃取至上清液所致。萃取包括液-液萃取(LLE, 萃取溶剂有二氯甲烷、氯仿和正己烷等)、固相萃取(SPE)、柱前提取、分散液-液微萃取和采用索氏提取器萃取等。预处理还包括微量渗析、超滤、微萃取和稀释等。Liang等在超滤过程中采用SDS和ACN的混合液作为置换溶液置换与蛋白质键合的LVFX、MXFX、TVFX和GTFX,以测定药物总量,发现该法回收率>95%。Xu等研究了基于ACN/水体系的LLE在预处理时的效果,结果表明,ACN和0.3%磷酸水溶液(70:30)效果最优。之所以用ACN和水溶液是为避免使用纯ACN时的样品固化问题,但ACN含量过低会导致蛋白质析出不完全。而70:30这一比例即可获得最优的回收率,又可去除蛋白质。然而,萃取和沉淀技术在血浆、血清、BAL、尿液和组织匀浆的预处理过程中并不常见。稀释是预处理药物制剂的常规方法。采用分离技术对LVFX进行分析时往往需要添加内标,内标的加入无疑为结果的可重复性和准确性提供了保障。
检测限(LOD)和定量限(LOQ)与所测样品及检测器息息相关,对荧光检测器而言,药物制剂的LOD和LOQ可低至10-9g/mL。检测器对血浆、尿液和血清的检测限更高,且MS的灵敏度高于UV和FLD。但在常规临床实践中,峰浓度和谷浓度往往mg/L级别上,故不需要检测极低的浓度。
2.2 巴洛沙星
为检测BLFX,HPLC常与MS和UV检测器联用(表2)。分离常用C18柱,有机相为ACN和甲醇,往往向水相中加入醋酸铵和磷酸二氢钾。流动相为弱酸性,当用磷酸二氢钾调pH至6.5时,样品的分离度和拖尾得以明显改善。Bian等评价了10 mmol/L醋酸铵在不同pH值(6.65 vs 3.0)时的效果,发现低pH值有利于改善样品的分离度和拖尾情况。
对血浆而言,预处理可用LLE,而药物制剂则可采用稀释。在LLE中,二氯甲烷和乙酸乙酯的混合液与正己烷和异丙醇的混合液相比效果更优。在萃取过程中,不推荐使用酸(盐酸/HCl, 1mol/L)或碱(氢氧化钠/NaOH, 1mol/L),这是由于酸和碱的存在会导致强干扰。MS的LOD和LOQ极低,且常选取准分子离子峰m/z 390 ([M+H]+)进行检测。
2.3 帕珠沙星
PZFX的检测方法有HPLC和CE技术,且二者可用于药物制剂(片剂)、牛奶和生物体液如血清、血浆、尿、肌肉匀浆、唾液和龈沟液等(表3)。HPLC常用C18和C8柱,且流动相中的ACN含量一般不超过15.5%,其它组分为0.5%磷酸和1% TFA或0.1%甲酸,并用TEA调pH至3.0。CE通常在室温下进行,背景电解质(BGE)为三羟甲基氨基甲烷(TRIS)和磷酸水溶液。β-环糊精和磷酸盐也可作为添加物,且pH需在5.04~9.00。所用电压取决于毛细管长度,毛细管越短电压越低。
样品预处理包括蛋白质沉淀(甲醇、ACN或6%的高氯酸甲醇溶液)、LLE(二氯甲烷)和稀释等。LOD取决于预处理,萃取为0.01~0.3 μg/mL,蛋白质沉淀为0.01~0.1 μg/mL,稀释尿液为7 μg/mL。
2.4 司帕沙星
SPFX的检测方法主要有光谱技术和色谱技术(表4),分离主要靠HPLC、超高效液相色谱(UPLC)和CE。研究发现,与UPLC相比,单引物扩增反应(SPAR)可降低拖尾效应和缩短保留时间(10倍)。UPLC的理论塔板数是HPLC的3倍,分离效果更优。色谱法的检测器有UV、DAD和MS。在MS/MS分析中,经常采用m/z 393.2→349.3和m/z 392.9→348.7的MRM模式。分离主要用C18柱,且可应用于血浆、血清、尿液和组织(肌肉)。流动相的有机相主要为ACN,其在等度洗脱时含量可达80%,但在其它条件下,其含量仅在12~20%。除ACN外,甲醇也较为常见。流动相中的添加物主要有0.1%磷酸、5%醋酸、磷酸二氢钠(NaH2PO4)、54 mmol/L甲酸、10 mmol/L醋酸铵和0.1%TFA等。CE在四硼酸缓冲液中进行,并向其中加入硅纳米粒,且pH在9.0左右。
样品预处理包括蛋白质沉淀(20%高氯酸和ACN)、LLE(乙酸乙酯)、用ACN/水外加磷酸和正己烷萃取及稀释。向流动相中加入少量的磷酸可提高肌肉组织的SPFX回收率,但加入甲酸时回收率较低。LOD取决于检测器,且MS最优。
3 第四代氟喹诺酮
常见的第四代氟喹诺酮有MXFX、TVFX、西他沙星(STFX)、普卢利沙星(PLFX)、吉米沙星(GMFX)和加替沙星(GTFX)等。其中,MXFX抗菌谱极广,包括革兰氏阳性菌和阴性菌如葡萄球菌、链球菌和肠球菌及非典型性细菌和厌氧菌等。MXFX可用于治疗结膜炎、角膜炎、术前和术后控制眼部感染、CAP和耐多药结核病(MDR-TB)的治疗。包含MXFX的疗法,可缩短治疗结核病(TB)的疗程。TVFX的抗菌谱包括革兰氏阳性菌和阴性菌,但本品由于会导致特异性肝毒性于1999年撤出市场,目前仅作兽药使用。STFX的抗菌谱包括革兰氏阳性菌和阴性菌、衣原体属和支原体属,对耐喹诺酮金葡球菌、肺炎球菌属和假单胞菌属同样有效。PLFX是尤利沙星(ULFX)的前药,其在体内可迅速的转化为后者。PLFX主要用于治疗单纯性膀胱炎、慢性支气管炎急性发作及儿童和成人下尿路感染。GMFX也是广谱抗菌喹诺酮,其对革兰氏阳性菌的活性显著增强。本品对肺炎链球菌的活性是MXFX的4倍,且对流感嗜血杆菌、卡他莫拉菌和非典型性嗜肺性军团病杆菌、衣原体属和支原体属同样有效。GTFX同样对革兰氏阳性菌和阴性菌具有良好的活性,其对耐甲氧西林肺炎链球菌也具有良好的活性。

表2 BLFX的检测方法
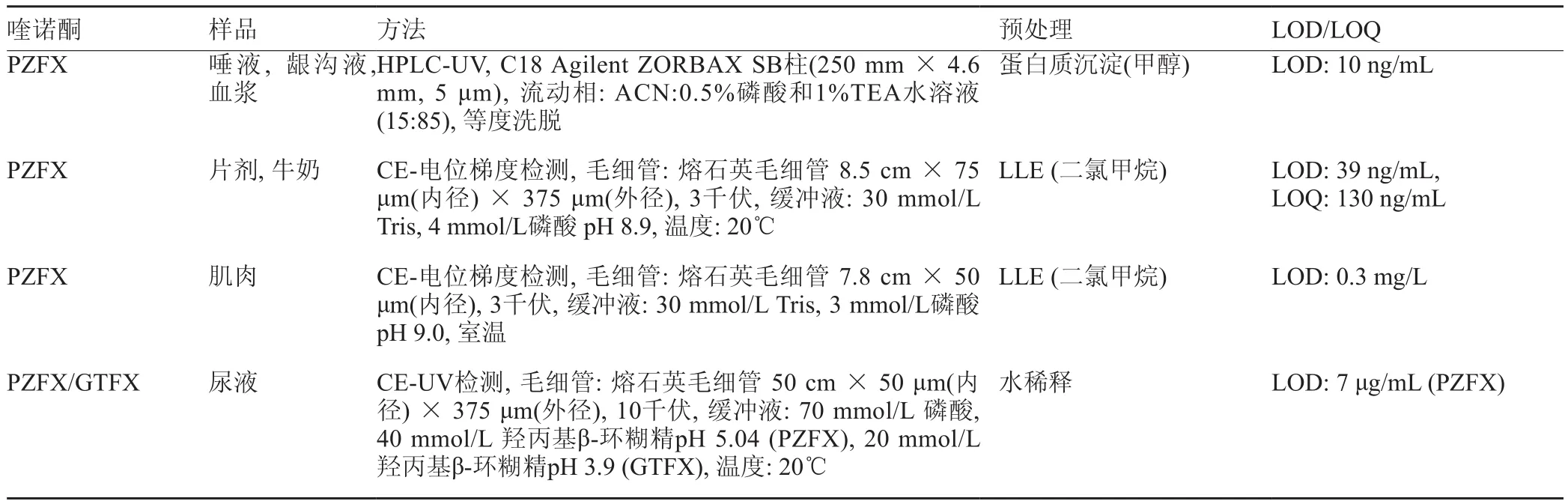
表3 PZFX的检测方法
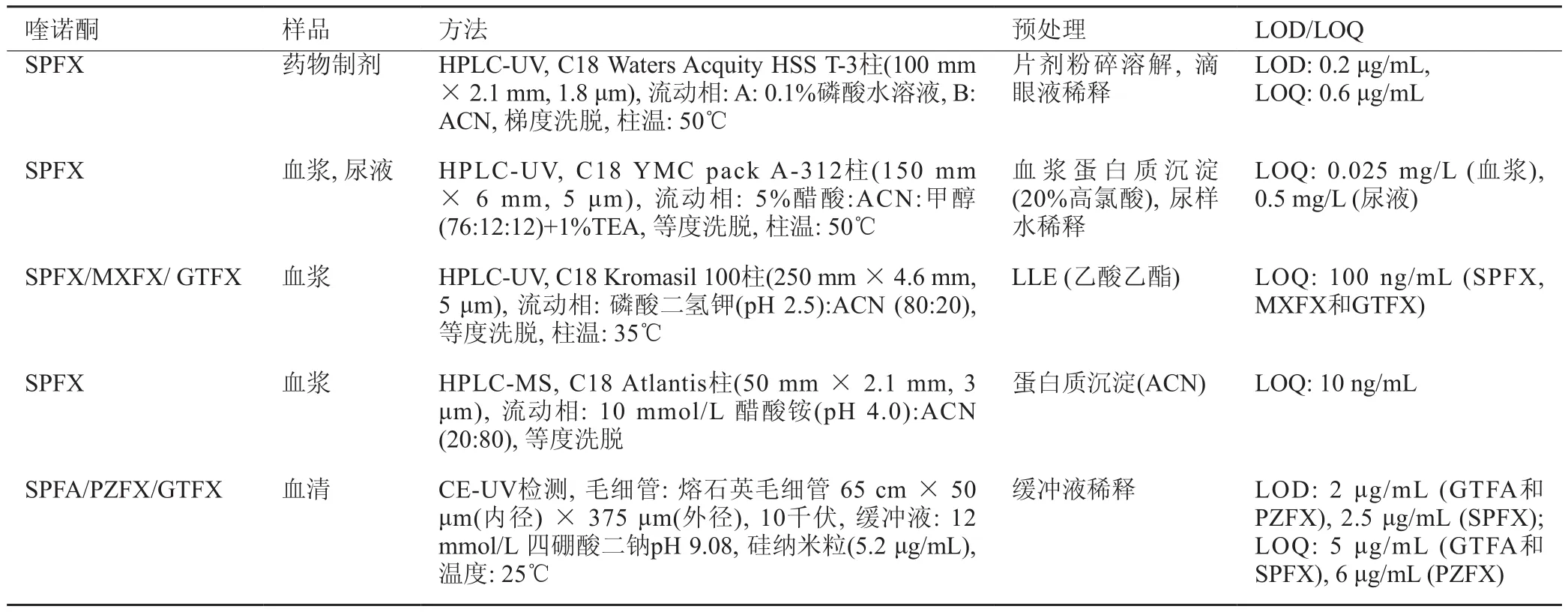
表4 SPFX的检测方法
3.1 莫西沙星
MXFX的分析方法众多,但主要是HPLC和各种检测器如UV、FLD、MS和DAD联用(表5)。UVVis可用于检测药物制剂或纯物质,而除此之外,CE也有文献报道。
HPLC的流动相主要为磷酸缓冲液,且浓度在10~50 mmol/L,但也有使用高浓度磷酸钠缓冲液(0.25 mol/L)的报道。其它添加物大多数为羧酸如柠檬酸、甲酸、醋酸、TFA或三氟乙酸酐,而盐主要为醋酸铵和SDS。Chan等发现TFA对荧光信号无干扰。离子对试剂也可用作添加剂,如0.03~2.00%TEA等。MS检测器所需浓度最低,而对其它检测器而言,TEA的最低浓度通常为0.1%。其它离子对试剂还有TBAA、TBAmBr和四丁基氯化铵(TBA·Cl)等,由于离子对试剂会和硅醇发生相互作用,会改善拖尾情况。离子对试剂总体而言会降低固定相的硅醇利用度,故用量尽可能少。添加离子对试剂过多时会导致柱平衡时间的延长和难以从柱子上冲洗干净,导致柱体污染。当pH在4.5和5.5时,拖尾严重,这可能是由于固定相硅醇的负电荷和MXFX胺基的正电荷共同作用的结果。但将pH降低至3.5时拖尾现象有所改善,这是由于硅醇在pH高于3.5时会发生离子化进而与伯胺和仲胺结合。水的含量在等度洗脱时通常为57~95%,而有机相可为ACN、甲醇或二者混合物,且pH为2.5~6。Laban-Djurdević等优化了响应面分析法,发现ACN的含量及pH是影响保留时间和分辨率的最重要因素,其次是磷酸缓冲液的离子强度。研究显示,响应面平面最大位置在ACN的含量为10~15%,pH为3.0~4.5。
在MS/MS分析中,经常采用m/z 402.1→260.0、m/z 402.0→358.2、m/z 402→384和m/z 402→358的MRM模式。
CE也可用于MXFX的分离,且BGE由缓冲液(有机相和无机相)、盐和TEA组成。检测在室温下进行,且γ-环糊精硫酸的加入可提高MXFX和其对映异构体的分离度。pH取决于流动相的组成,且酸和碱均可。
Ashour等报道了一种在四价铈离子(Ce4+)存在的条件下将MXFX与3-甲基-2-苯并噻唑啉酮腙盐酸盐单水合物(MBTH)络合以测定药物制剂和纯物质中MXFX的简单方法,该法适用于动力学分析。Djurdevic和Cruz等分别采用基于HPLC和CE的方法进行杂质分析,HPLC适用于杂质和强制降解产物分析,而CE适用于检测MXFX的S,S-、R,R-、R,S-和S,R-光学异构体。其中,S,S-是MXFX的活性构型,其它3个则为潜在的MXFX强制降解产物。
样品预处理方法有萃取(经典的LLE, 用二氯甲烷、乙酸乙酯; SPE; 在线柱前萃取和氰基丙咪嗪萃取)、蛋白质沉淀(ACN、甲醇、二者的混合液和高氯酸)、稀释和过滤等。
可分析的样品有血浆、血清、血液、唾液、玻璃体和房水、药物制剂(滴眼液和片剂)及纯物质等。在生物样品中,用到的主要预处理方法为萃取和蛋白质沉淀。但以SDS为替换试剂的超滤或微过滤技术可增强蛋白质的溶解性和降低蛋白质与药物的结合,故二者也偶有使用。对SDS浓度优化结果表明,SDS最优浓度为10 mmol/L且需磷酸缓冲液调pH至3.0。进一步研究发现,SDS的加入可提高荧光强度,有利于分离。药物制剂的预处理主要是稀释,且在绝大多数情况下需加内标。
LOD和LOQ大多数集中在μg/mL级别,但MS和FLD或UV检测器可检测到ng/mL水平。
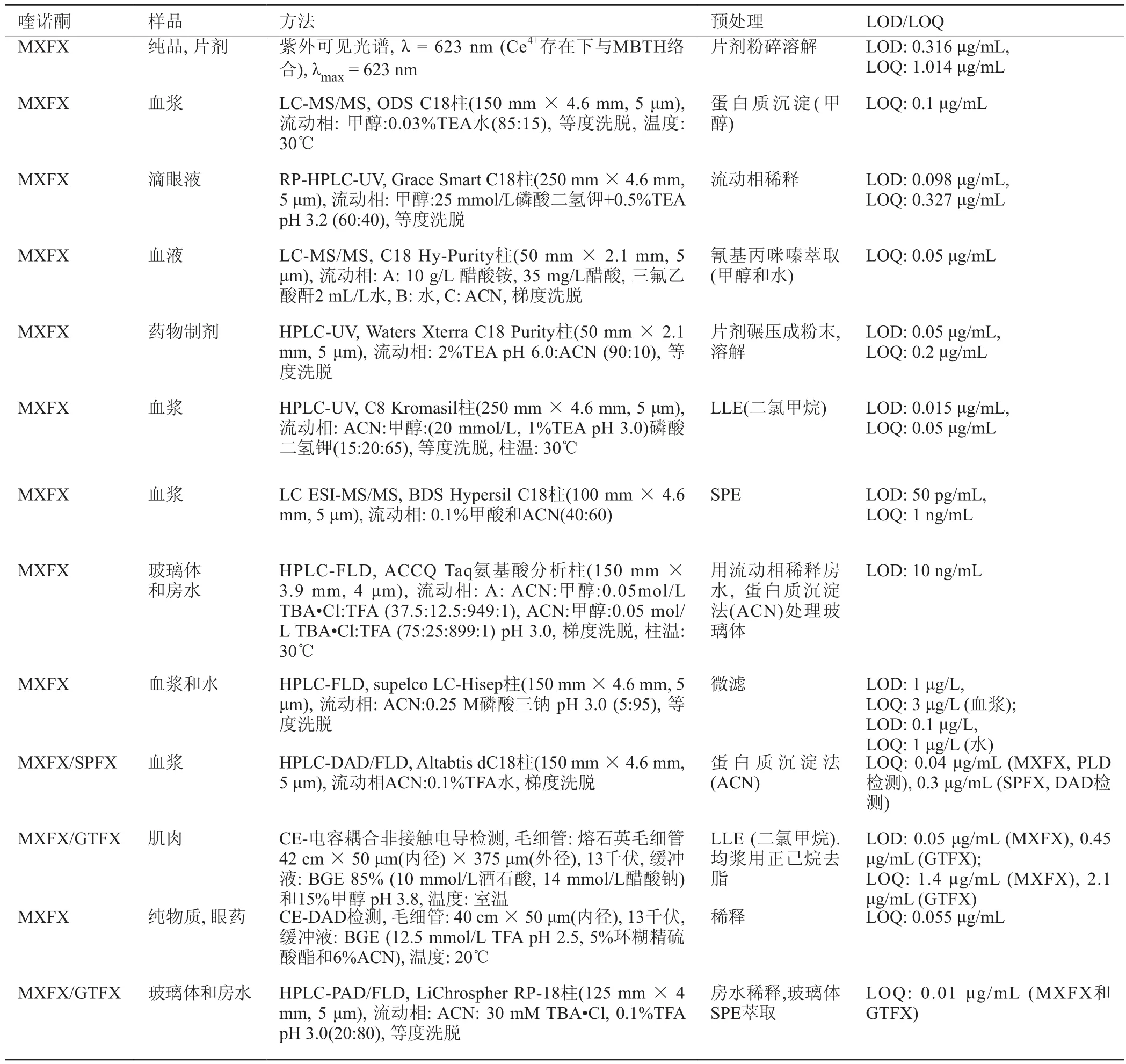
表5 MXFX的检测方法
3.2 曲伐沙星
TVFX可用HPLC与UV或FLD联用进行分析(表6),分离用C18柱,有机相为ACN或与甲醇的混合物。离子对试剂可用四丁基氢氧化铵、四丁基铵的硫酸氢盐、醋酸盐或TFA盐,无机试剂可用磷酸二氢钠、磷酸钠或0.1%甲酸。其它添加物可用柠檬酸或SDS,流动相需弱酸性。可分析的样品主要有尿和血浆。对血浆和痰样品而言,可用ACN、ACN与高氯酸混合液和20%高氯酸预处理样品,也可用0.5%SDS超滤样品,而尿样需要稀释。蛋白质沉淀法的LOD与上述色谱分离相似,而尿样的LOD较高。TVFX也可用微分脉冲吸附溶出伏安法进行分析,此时,预处理为过滤,且LOD高于其它方法。
3.3 西他沙星
STFX可用色谱法与MS联用进行分析(表7)。在MS/MS分析中,经常采用m/z 410.1→392.1和m/z 410.1→392.2的MRM模式。所报道的流动相相似,即甲醇和0.1%的甲酸混合液。当提高甲酸的浓度时,需提高柱温。预处理用蛋白质沉淀法,试剂为甲醇和0.1%甲酸或异丙醇。
3.4 普卢利沙星
PLFX是ULFX的前药,二者往往同时检测,且ULFX通常被认为是PLFX的杂质(表8)。PLFX和ULFX均可以HPLC与UV或MS检测器联合或CE法直接检测。在MS/MS分析中,经常采用m/z 462→444、m/z 462→418、m/z 460→360和m/z 462→350的MRM模式分析PLFX,采用m/z 350→248的MRM模式分析ULFX。目前,用于检测的有片剂、降解产物、房水、血浆和尿液,所用柱子有C18、C8和亲水作用色谱(HILIC)柱。流动相中有机相主要为ACN和甲醇,ACN可被醇替代,但用HILIC柱要达到同样的保留时间需提高有机相的含量。当用MS检测时,流动相还可加入磷酸二氢钾、醋酸铵、磷酸和甲酸等添加物。而CE法中的BGE则由柠檬酸钠、柠檬酸和硫酸钠。
对于片剂而言,预处理主要是粉碎和用适宜的溶剂溶解,液体样品则可稀释,蛋白质沉淀则可用甲醇。LOD取决于检测样品和检测器,其中MS对PLFX而言LOD最低。同样的检测手段,ULFX的LOD要高于PLFX。
3.5 吉米沙星
GMFX可用HPLC和高效薄层色谱(HPTLC)分析,其中HPLC可与FLD、UV和PDA检测器联用,而HPTLC则与UV检测器联用。常用的柱子有C18和HILIC柱,有机流动相有ACN和甲醇,水相为10 mmol/L的醋酸铵或0.1%TFA或醋酸钠或磷酸水溶液。HPTLC则用乙酸乙酯和氨水做展开剂。样品可以是血液、血浆和反应液。预处理的方法包括萃取、超滤、LLE和稀释。FLD的LOD要低于UV和PDA检测。
3.6 加替沙星
GTFX可用HPLC和CE分离,其中HPLC可与FLD、UV和MS检测器联用,而CE则与UV、电容耦合非接触性电导检测和电致发光检测器联用(表9)。

表6 TVFX的检测方法

表7 STFX的检测方法
柱色谱主要用C18柱,有机流动相有ACN和甲醇,离子对试剂有TBAA、TEA、TBAmBr和TBA·Cl。其它添加剂有磷酸盐、SDS、柠檬酸、0.1%甲酸、TFA、醋酸铵和磷酸。流动相微酸性,可等度洗脱也可梯度洗脱。除少数HPLC在35和28℃进行外,其它HPLC均可在室温条件下进行。CE法可在PBS、TRIS/盐酸、四硼酸缓冲溶液、环糊精的磷酸酯缓冲溶液、四硼酸二钠盐与硅纳米粒、酒石酸和醋酸钠体系进行。在MS/MS分析中,经常采用m/z 375.9→332.0和m/z 375.9→260.9的MRM模式进行分析。
可用于分析的样品有片剂、药物制剂、血清、血浆、血液、尿样、肌肉、房水和玻璃体及食物样品。预处理方法包括碾碎和溶解、超滤、在线柱萃取、蛋白质沉淀(ACN、甲醇或二者混合物)、用合适溶剂溶解、LLE、SPE和加速溶剂萃取(ASE)。Fu等发现SPE在分析过程中不会产生不可接受的干扰,而Tasso等则研究了在线SPE和HPLC联用技术。LOD和LOQ均取决于所用检测器,且MS较低。
4 结束语
氟喹诺酮含有两个可与质子结合的基团即氨基和羧基,其两性离子的特性使得氟喹诺酮很容易离子化,这就导致对此类物质的分析显得较为复杂。其中,峰拖尾和分离度差是分析过程中遇到的主要问题。ACN和甲醇是梯度洗脱和等度洗脱最常用的有机溶剂,且ACN的洗脱能力高于甲醇,使得色谱图上的峰信息较全。向ACN中加入甲醇,对分离度有影响。分离氟喹诺酮时经常用反相,且常用的柱子有C18、C8和C4柱。HILIC柱也偶有使用,其是RP-HPLC分离亲水离子样品的良好替代品。HILIC需要至少80%的有机相(主要是ACN),且适合与MS联用。在氟喹诺酮的分析中,一个重要的组分是离子对试剂(TEA, TBAA, SAS或其它)。当加入离子对试剂时可能会导致柱平衡时间延长和试剂残留,但离子对试剂可加强药物与固定相的相互作用。因此,是否需要加入离子对试剂,检测人员需综合考虑。所用色谱柱和其它添加剂如有机或无机盐、离子对试剂,及pH均可能影响峰型和LOD及LOQ。其它需要考虑的是样品的预处理和所用检测器,生物流体的LOD或LOQ要高于药物制剂或溶液。蛋白质沉淀的LOD和LOQ要高于萃取(LLE或SPE)。尽管萃取相对费力,但在检测低含量样品时不可或缺。对药物制剂而言,预处理稀释就足够了,且可用UV、可见光和荧光光谱检测。对于需要去蛋白质的样品,蛋白质沉淀法是最简单的途径,且回收率高于萃取(LLE和SPE)。LLE或SPE相对较为复杂,需要蒸除溶剂和溶解样品,这不仅费时,而且需要额外的试剂和仪器。组织匀浆的预处理更为复杂,需要去除蛋白质和脂质。蛋白质沉淀需要在溶剂中完成,且需要加水溶液,以避免蛋白质固化包裹样品,导致回收率降低。当样品中含有不止一个能被检测出的组分时,需运用分离技术。当LOD和LOQ较低时,需采用荧光检测器或MS检测器。MS检测器与萃取联用时,LOD可低至pg/mL。MS分析需要有机酸作为质子化溶剂,如甲酸、乙酸或与甲酸铵、醋酸铵的混合物。串联MS/MS是MS检测中使用最多检测手段。
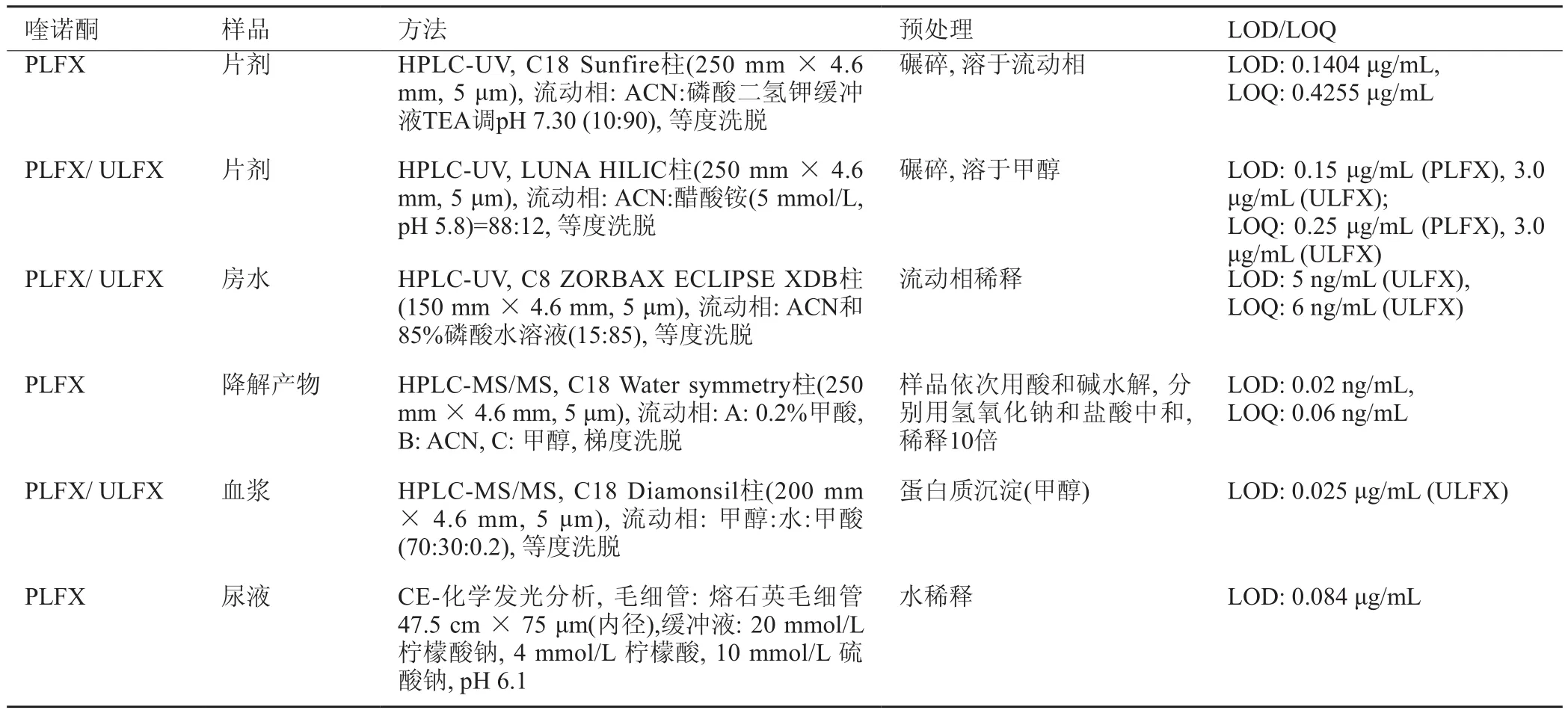
表8 PLFX和ULFX的检测方法
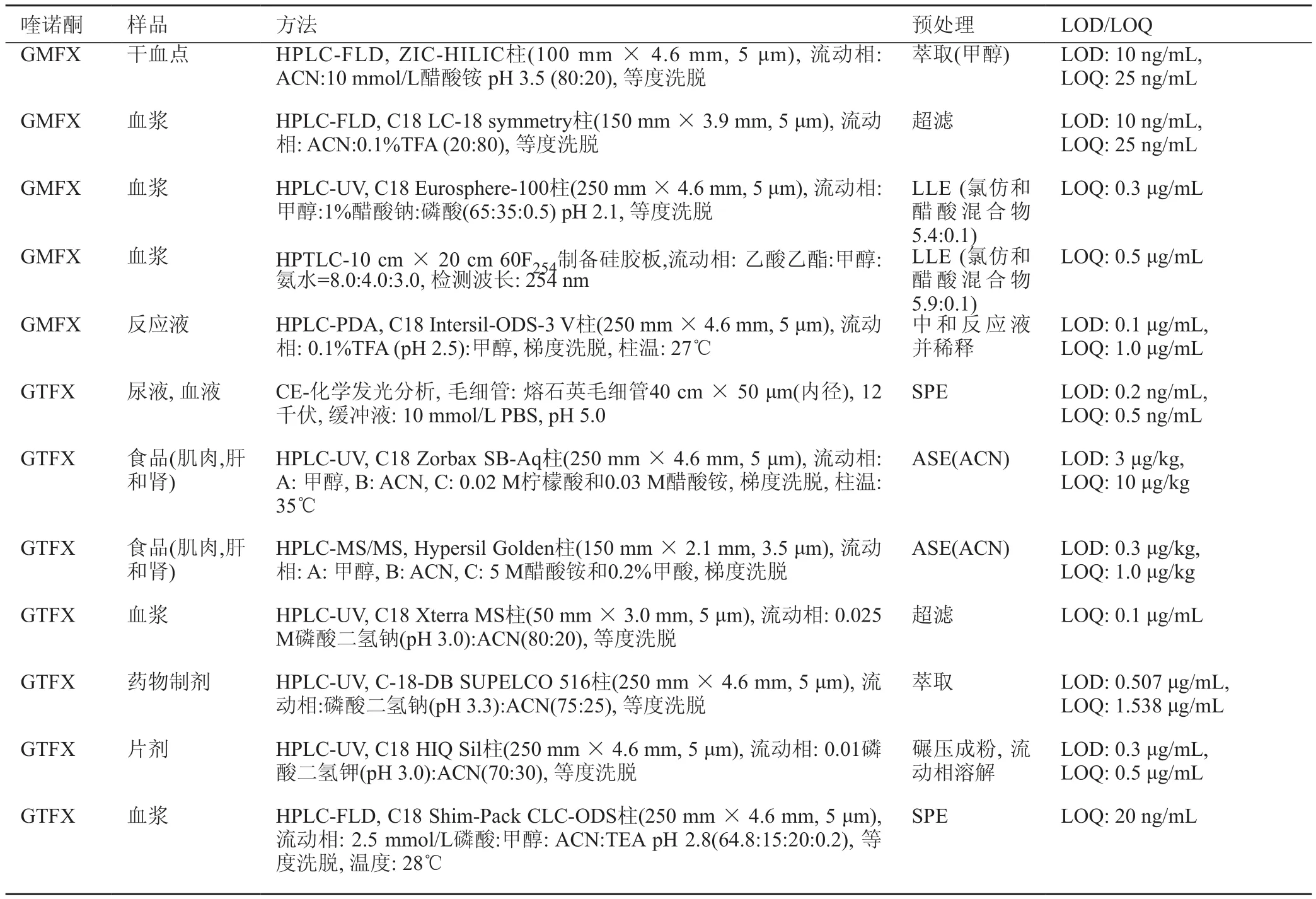
表9 GMFX和GTFX的检测方法
在临床分析中,需要快速的获得分析结果以确定治疗方案,蛋白质沉淀和HPLC联用是理想的搭配。若没有MS检测条件,采用内标法的FLD或UV检测也不失为一种快速、廉价的替代方案。第三代和第四代氟喹诺酮在血液和其它流体的浓度在mg/L水平,HPLC与FLD或UV联用恰在此范围内。而用内标法可补偿萃取过程中的损失,可重复性较高。
总之,本文介绍近年来所发展的第三代和第四代喹诺酮的分析方法及样品的预处理方法,为分析人员今后从事相关研究提供参考。
