喹啉和喹诺酮二聚体的生物活性
2019-10-12魏增全冯连顺编写刘明亮郭慧元审校
魏增全,冯连顺 编写 刘明亮,郭慧元 审校
(1 天津大学化工学院,天津,300072
2 天津红日药业股份有限公司,天津,301700
3 中国医学科学院医药生物技术研究所,北京 100050)
1 前言
喹啉和喹诺酮结构片段广泛存在于天然产物中,其衍生物具有抗菌、抗炎、抗肿瘤、抗丙肝(HCV)、抗结核(TB)、抗疟疾、抗人类免疫缺陷病毒(HIV)和抗阿尔茨海默病(AD)等多种生物活性,在临床上有着广泛的应用前景。
与相应的单核化合物相比,双核化合物往往显示出某些特殊的性质如生物活性增强、抗菌谱更广等。喹啉和喹诺酮二聚体也具有多种生物活性,且其中的某些化合物如喹哌等已成功应用于临床,故喹啉和喹诺酮二聚体(见图1)引起了药物化学家的极大兴趣。
在过去的30年中,药物化学家设计、合成并评价了众多喹啉和喹诺酮二聚体的体内外生物活性,发现了若干具有苗头的候选物。本文将重点介绍喹啉和喹诺酮二聚体在抗菌、抗肿瘤、抗疟疾和抗TB领域的最新研究进展,并归纳这类化合物的构-效关系(SAR),以期指导药物化学家更合理的设计此类化合物。
2 抗菌活性
细菌感染是导致院内感染的罪魁祸首,给各国医疗系统带来了沉重的负担。更为严峻的是,细菌对几乎所有类型的抗生素均产生了不同程度的耐药性,使得抗生素的药效呈不断下降之势。据估计,每年因耐药菌导致的死亡人数高达70万,而若无法有效阻止这一趋势,至2050年死亡人数可能会急剧增长到1000万。显然,研发对药敏型和耐药型致病菌均有效的新型抗生素势在必行。
喹啉和喹诺酮衍生物尤其是氟喹诺酮目前在临床上广泛用于各种细菌感染的治疗,不幸的是,细菌对这类药物也产生了耐药性。研究表明,适当的增大喹啉和喹诺酮衍生物的分子量或向其母核引入大取代基并不会影响渗透性,故向喹啉和喹诺酮母核再引入一个喹啉和喹诺酮结构片段的二聚体策略可能会获得活性更高的候选物。
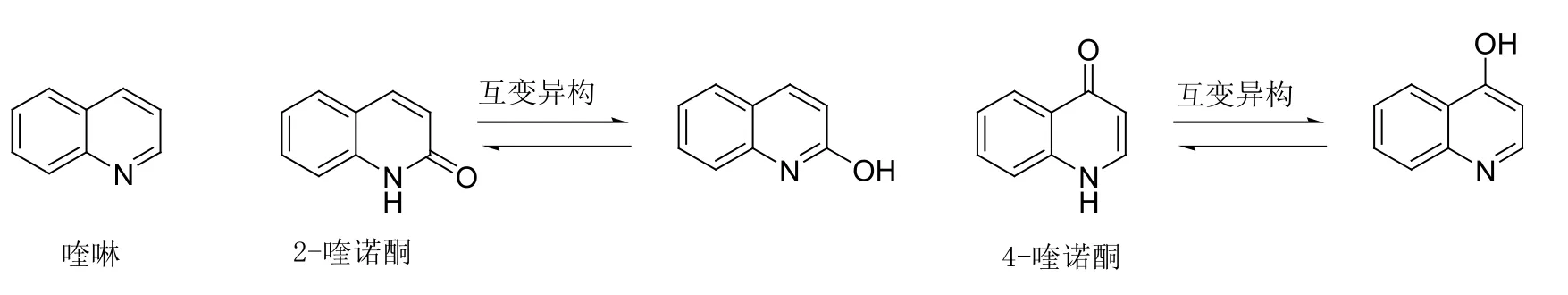
图1 喹啉、2-喹诺酮和4-喹诺酮的化学结构
药代动力学研究结果表明,对3-羧酸喹诺酮而言,N-1位为芳基时可提高此类化合物的生物利用度。基于此,Chepyala等评价了一系列以次乙基和1,2-环己二基为连接子的喹诺酮二聚体1(见图2)的体外抗大肠埃希菌和金黄色葡萄球菌(金葡球菌)活性。结果表明,所有二聚体(在浓度为50和100 μg/disc时对大肠埃希菌和金葡球菌的抑菌圈直径为0~22 mm)的活性均弱于对照药环丙沙星(在浓度为50和100 μg/disc时对大肠埃希菌和金葡球菌的抑菌圈直径为36和38 mm),且超过半数的二聚体对金葡球菌未显示出任何活性。SAR研究结果显示,羧酸衍生物(R =H)的活性优于相应的乙酯,且向C-8位(R2)引入氟原子可在一定程度上提高抗菌活性。
以氨基酸为连接子的喹诺酮(萘啶酸/恶喹酸)-氟喹诺酮(环丙沙星/诺氟沙星)二聚体2和3可能会降低抗生素的降解率,提高其在作用靶点的浓度,进而提高抗菌活性。抗菌评价结果表明,二聚体2和3对所测得金葡球菌、化脓性链球菌、伤寒沙门氏杆菌和铜绿假单胞菌具有弱到中等强度的活性,最小抑制浓度(MIC)为3.3~2116.5 μmol/L,但大多数二聚体弱于相应的母药。尽管如此,化合物2b (MIC:7.8 μmol/L)和2f (MIC: 3.3 μmol/L)对革兰阳性菌金葡球菌和化脓性链球菌具有潜在的活性,而母药萘啶酸、恶喹酸、环丙沙星和诺氟沙星(MIC: 74.6~3914.3 μmol/L)的活性则相对较弱。此外,化合物3g,h(MIC: 7.6和7.4 μmol/L)对伤寒沙门杆菌的活性也不亚于4个母药(MIC: 7.2~10.3 μmol/L)。
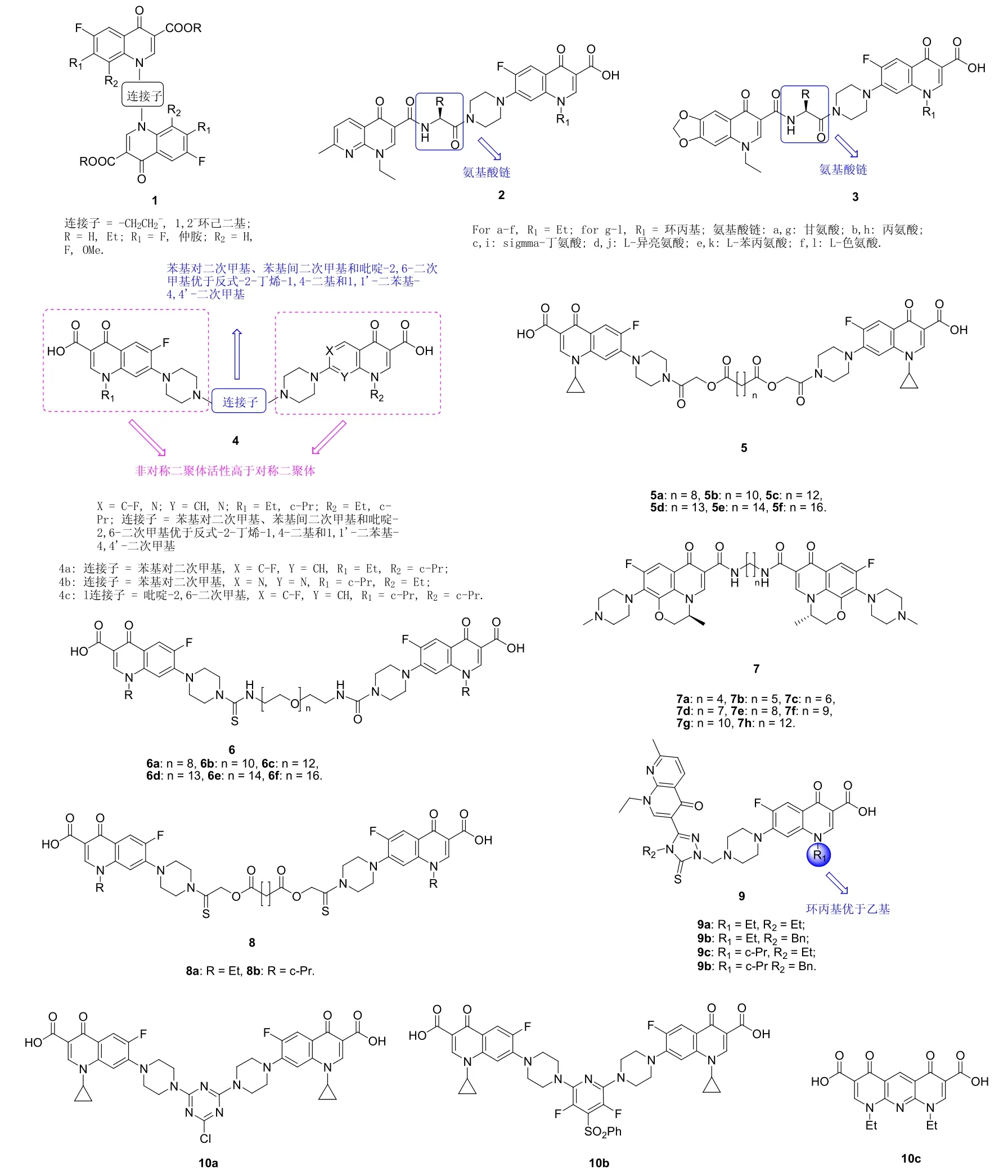
图2 喹诺酮二聚体1~10的化学结构
对一系列对称的环丙沙星、诺氟沙星和吡哌酸二聚体及不对称环丙沙星-诺氟沙星和环丙沙星-吡哌酸二聚体的体外抗耐药菌金葡球菌 SA 1199 (耐氟喹诺酮野生型细菌)、SA 1199-3 (实验室培育的诱导norA过表达且DNA促旋酶或拓扑异构酶IV未突变的SA 1199突变菌株)、SA 1199B (组成性过表达norA且对氟喹诺酮具有一定耐药性的SA 1199变种)、耐甲氧西林金葡球菌(MRSA)临床分离株和耐万古霉素金葡球菌(GISA)活性测试结果表明,与母药环丙沙星、诺氟沙星和吡哌酸(MIC: 0.125~>16 μg/mL)相比,所有二聚体(MIC: <0.03~1 μg/mL)对所测的金葡球菌SA 1199、SA 1199-3、SA 1199B和MRSA活性均有所提高。不仅如此,某些二聚体对GISA也具有中等强度的活性,MIC为 2~8 μg/mL,而母药(MIC: >16 μg/mL)未显示出任何活性。以上结果表明,喹诺酮母核和连接子对活性均有较大影响。总体而言,不对称二聚体的活性优于对称二聚体。对对称的二聚体而言,环丙沙星二聚体>诺氟沙星二聚体>吡哌酸二聚体。对连接子而言,苯基对二次甲基、苯基间二次甲基和吡啶-2,6-二次甲基优于反式-2-丁烯-1,4-二基和1,1'-二苯基-4,4'-二次甲基。其中,二聚体4a-c(MIC: <0.03~0.125 μg/mL)不仅对金葡球菌SA 1199、SA 1199-3、SA 1199B和MRSA的活性是环丙沙星、诺氟沙星和吡哌酸的≥4倍,而且对GISA也具有良好的活性(MIC: 4, 2和8 μg/mL)。进一步研究发现,这类二聚体对药敏型和耐药型肺炎链球菌也具有优秀的活性。与环丙沙星(作用于拓扑异构酶IV)不同的是,这类二聚体可通过作用于肺炎链球菌和金葡球菌的DNA促旋酶发挥功效,作用靶点的转变可能是这类化合物活性更高的重要原因。值得一提的是,这些二聚体的体外抗化脓性链球菌、结核分枝杆菌(MTB) H37Rv、大肠埃希菌、耐万古霉素表皮球菌(VRE)和铜绿假单胞菌活性不亚于母药,提示这类化合物具有广谱抗菌活性,值得深入研究。
Azéma等评价了通过C-7位连接的环丙沙星二聚体5,6和通过C-6位连接的左氧氟沙星二聚体7的抗肿瘤、抗菌和抗分枝杆菌活性,发现此类二聚体可特异性的抑制金葡球菌和肿瘤细胞的生长,但对MTB无明显活性。环丙沙星二聚体5e和6e对所测所有肿瘤细胞的半抑制浓度(IC50)分别为3~8 μmol/L和0.1~9 μmol/L,活性分别是母药环丙沙星(IC50: 89~476 μmol/L)的15~40和32~890倍,而左氧氟沙星二聚体7f (IC50: 0.2~0.7 μmol/L)的抗肿瘤活性则是母药左氧氟沙星(IC50: 67~622 μmol/L)的100~1240倍。进一步分析发现,这些二聚体可克服某些肿瘤细胞系的天然耐药性,这可能归因于此类二聚体可通过激发肿瘤细胞的凋亡性或非凋亡性细胞死亡过程。左氧氟沙星二聚体7的抗革兰阴性菌(铜绿假单胞菌和大肠埃希菌)活性优于环丙沙星二聚体5,6,但后者的抗金葡球菌活性更高。其中,环丙沙星二聚体5b-e和6b-d不仅对药敏型金葡球菌的活性极高(MIC: ≤0.97 μmol/L),而且对所测的2株MRSA和耐多药临床分离株也具有潜在的活性,MIC为0.0081~125 μmol/L。特别值得一提的是,二聚体6d对所测2株MRSA的MIC低至0.06 μmol/L,对所测2株耐多药金葡球菌临床分离株的MIC为1.9和15.6 μmol/L,具有治疗MRSA和耐多药金葡球菌感染的潜力。聚乙二醇硫酰胺连接的诺氟沙星和环丙沙星二聚体8a,b (MIC: 3.9~>125.4 μmol/L)对所测绝大多数菌株的活性弱于母药诺氟沙星和环丙沙星(MIC: 1.3~>6.2 μmol/L),提示连接子对二聚体的活性至关重要。
氟喹诺酮-1,2,4-三氮唑-5(4H)-硫酮杂合体具有优秀的抗菌活性,故将1,2,4-三氮唑-5(4H)-硫酮嵌入至氟喹诺酮二聚体也可能会获得抗菌活性较高的候选物。1,2,4-三氮唑-5(4H)-硫酮连接的萘啶酸-诺氟沙星/环丙沙星衍生物9对大肠埃希菌、假结核耶尔森氏菌、铜绿假单胞菌、金葡球菌、粪肠球菌、蜡样芽孢杆菌和耻垢分枝杆菌显示出一定的活性,MIC为<1~78 μg/mL。SAR研究结果表明,萘啶酸-环丙沙星衍生物9c,d (MIC: <1~9.7 μg/mL)的活性优于相应的萘啶酸-诺氟沙星衍生物9a,b和对照有氨苄西林(MIC:10~>128 μg/mL)。
均三嗪连接的环丙沙星二聚体10a及其衍生物10b (MIC: 0.25~2 μg/mL)具有广谱抗菌活性,二者对金葡球菌、肠球菌属、大肠埃希菌、变形杆菌、志贺氏菌、克雷伯菌、假单胞菌和肺炎克雷伯菌的活性总体上不亚于母药环丙沙星(MIC: <0.125~1 μg/mL),但二聚体10c则对所测得革兰阳性菌和阴性菌均未显示出任何活性。
向药物分子中嵌入金属离子往往会深刻影响药物的生物活性,且某些金属化合物如二茂铁喹已用于临床或处于临床评价阶段,故金属螯合物引起了研究人员的极大兴趣。研究显示,金属离子与喹诺酮螯合是这类药物发挥抗菌活性的关键步骤。显然,喹诺酮金属螯合物的抗菌活性可能优于喹诺酮自身。
据报道,低分子量的二价铜(Cu2+)螯合物对多种疾病有效,故将两个喹诺酮母核通过Cu2+连接的策略是该领域的研究热点。Cu(恩诺沙星)2(H2O)11(MIC:0.125~4 μg/mL)对所测金葡球菌、大肠埃希菌和铜绿假单胞菌的活性优于相应的单恩诺沙星-Cu2+螯合物(MIC: 0.5~4 μg/mL)和母药恩诺沙星(MIC: 1~8 μg/mL)。值得一提的是,与母药恩诺沙星(MIC: 1 μg/mL)相比,双恩诺沙星-Cu2+螯合物11 (MIC: 0.125 μg/mL,见图3)的抗革兰阴性菌活性提高了8倍。
加替沙星-金属螯合物12对所测包括耐药菌MRSA在内的革兰阳性菌显示出良好的活性,且几乎所有的螯合物的抗菌活性与对照药加替沙星、司帕沙星和吉米沙星相当或更优。
大多数二价铂(Pt2+)连接的氧氟沙星、培氟沙星、加替沙星、诺氟沙星、左氧氟沙星和司帕沙星二聚体的抗菌活性优于相应的母药,且活性与氟喹诺酮母核息息相关:司帕沙星 > 培氟沙星 ≈ 左氧氟沙星 > 氧氟沙星 > 诺氟沙星 > 加替沙星。其中,双司帕沙星-Pt2+螯合物13抗革兰阳性菌和阴性菌的MIC低至0.2~0.5 μmol/L,活性是上述6种氟喹诺对照有的2~25.5倍,值得进一步研究。
除上述双氟喹诺酮金属螯合物外,药物化学家还评价了若干其他双氟喹诺酮金属螯合物的抗菌活性。尽管某些螯合物的活性优于母药,但大多数螯合物的活性弱于母药。
3 抗肿瘤活性
肿瘤几乎可在人体各个器官增殖,是仅次于系血管疾病的第二大致死性疾病。其中,肺癌、前列腺癌、直肠癌、胃癌和肝癌是男性最为常见的癌症,而乳腺癌、直肠癌、肺癌、宫颈癌和胃癌则是女性多发癌症。据世界卫生组织(WHO)估计,每年约有1400万人罹患癌症, 其中约880万人因此丧命。仅2010年一年,全球因癌症而导致的年度经济损失高达1.16万亿美元。癌症在全球范围内的广泛传播和耐药癌症的不断涌现,使得研发新高效型抗癌症药物迫在眉睫。目前,尽管多种抗癌候选物处于临床研究阶段,但仍不能满足患者需求。因此,有必要研发新型抗癌药物。
喹啉和喹诺酮类化合物也具有潜在的抗肿瘤活性,其中的某些化合物如托泊替康(见图4)、Voreloxin和Quarfloxin已用于临床治疗各种癌症或处于临床评价阶段。值得一提的是,在I期临床试验中,喹啉二聚物Dactolisib对各种实体瘤有良好的疗效,目前正处于深入评价阶段,结果值得期待。因此,喹啉和喹诺酮衍生物是潜在的抗癌药物。

图3 双喹诺酮-金属螯合物11~13的化学结构
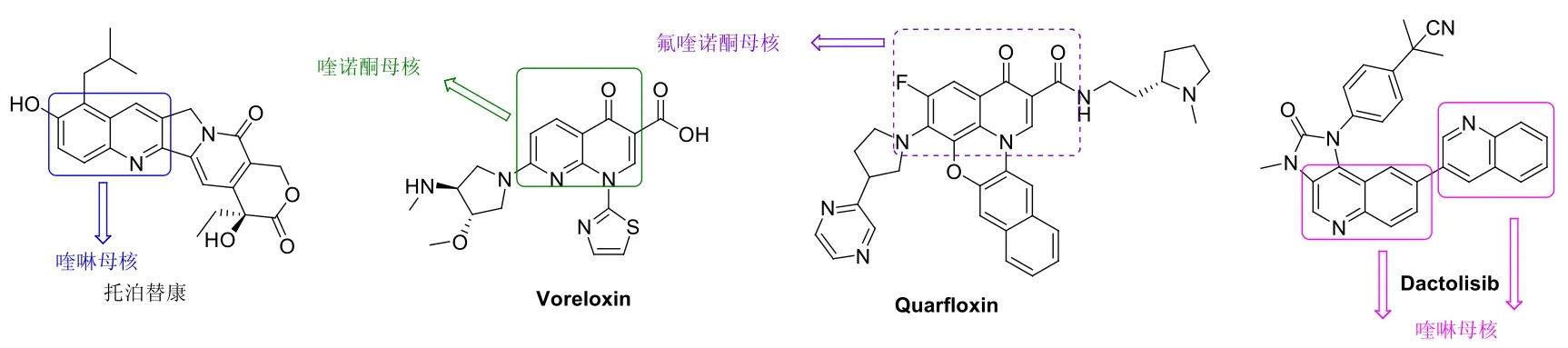
图4 托泊替康, Voreloxin, Quarf l oxin和Dactolisib的化学结构
对4-氧代-3-氟苯胺连接的喹啉二聚体14(见图5)和15的体外抗肿瘤(H460, HT-29, MKN-45,U87MG和SMMC-7721细胞系)研究结果表明,这类二聚体(IC50: 0.011~3.56 μmol/L)具有中等到良好的抗肿瘤活性。此类二聚体对不同的肿瘤细胞系活性迥异,且大多数化合物对H460和MKN-45细胞系的活性较高。SAR显示,R1位的取代基对活性影响不大,而R3位的取代基对活性有显著影响,且单吸电子基对活性有利,而双吸电子基和供电子基则对活性不利。值得一提的是,R3取代基的位置与活性密切相关,且间位优于对位。与二聚体14相比,化合物15的活性更高,提示向R2位引入氟原子可增强活性。其中,二聚体14d, 14e, 14m, 14n, 15a和15i (IC50:0.011~0.92 μmol/L)对所测所有肿瘤细胞系的活性均优于对照药 foretinib (IC50: 0.031~1.04 μmol/L),且对c-Met具有极强的抑制活性(IC50: 1.32~3.45 nmol/L),提示c-Met可能是这类二聚体的作用靶点。代表物14e的抑c-Met活性与foretinib相当(IC50分别为1.32和1.16 nmol/L),对所测5株肿瘤细胞系的IC50低至0.011~0.15 μmol/L,活性是foretinib的2.9~10.9倍,值得深入研究。进一步研究发现,喹啉核可被2-喹诺酮(16)取代,但不能被2-萘啶酮代替。代表物2-喹诺酮-喹啉16a (IC50: 0.031~0.52 μmol/L)对所测肿瘤细胞系的活性极高,对H460、HT-29和U87MG细胞系的活性分别是对照药foretinib的6.1、2.4和2.1倍。化合物16a对c-Met (IC50: 1.21 nmol/L)和Flt-3 (IC50: 2.15 nmol/L)均具有良好的抑制活性,但对c-Kit, VEGFR-2、PDGFR-b和EGFR的抑制活性(IC50:362.8~>100,000 nmol/L)较弱,提示c-Met和Flt-3是其主要作用靶点。
羟基喹啉二聚体17对KB3细胞系具有良好的活性,CC50为1.3~123 nmol/L,但弱于对照药多西他赛(CC50: 0.25 nmol/L)。SAR显示,与化合物17a (CC50:15 nmol/L)相比,向苯环的对位引入甲基(17b, CC50:2.6 nmol/L)和三氟甲基(17c, CC50: 1.3 nmol/L)可提高活性,而向对位引入硝基(17d, CC50: 42 nmol/L)或向邻位引入三氟甲基(17e, CC50: 123 nmol/L)则对活性不利。作者进一步测试了二聚体 17b,c对多株肿瘤细胞系和2株非肿瘤细胞系的活性,发现二者对绝大多数细胞系具有良好的活性,CC50为0.8~8.0 nmol/L。有趣的是,二聚体17c对所测所有细胞系的活性均优于化合物17b。作用机制研究结果显示,二者既不抑制微管蛋白也不抑制蛋白酶体,但对肿瘤坏死因子相关细胞凋亡诱导配体具有促凋亡作用。
喹啉二聚体18a-e具有良好的抑类端粒沉默干扰体1 (DOT1L)活性,IC50为1.06~21.67 μmol/L。SAR显示,这类二聚体的抑DOT1L活性与R位的取代基息息相关,且-NH2(18a, IC50: 1.5 μmol/L) > -OH (18b,IC50: 4.35 μmol/L) >> -OMe (18c, IC50: 21.67 μmol/L),提示氢键供体对活性有利。二聚体18d (IC50: 1.08 μmol/L)的活性优于18a,提示嘧啶环上的甲基对活性不利。化合物18e (IC50: 1.06 μmol/L)的抑DOT1L活性略优于其位置异构体18d,但其(IC50: 30.54和45.75 μmol/L)抗混合系白血病(MLL)表达急性白血病细胞MV4-11和非MLL重排白血病细胞活性较弱,这可能是由于该化合物的渗透性较差所致。

图5 喹啉二聚体和喹啉-喹诺酮衍生物14~19的化学结构
除上述喹啉二聚体外,某些其他二聚体也具有潜在的抗肿瘤活性,如喹啉二聚体19对DMAMB-468和MCF-7肿瘤细胞系的GI50分别为7.35和14.80 μmol/L,优于相应的单喹啉衍生物(GI50:8.22~51.57 μmol/L),值得进一步研究。
[1,2,4]三唑[3,4-b][1,3,4]噻二唑连接的氟喹诺-氟喹诺酮二聚体20(见图6)和21对所测得L1210、CHO和HL60肿瘤细胞系具有潜在的活性,IC50为0.12~26.2 μmol/L。其中,二聚体20a和21d (IC50:0.54和0.12 μmol/L)对HL60的活性最高,提示N-1位的环丙基为高活性所必需。进一步研究显示,用2,5-[1,3,4]噁二唑取代[1,2,4]三唑[3,4-b][1,3,4]噻二唑并不能提高抗肿瘤活性,而2-喹诺酮二聚体则仅显示出较弱的抗肿瘤活性。
4 抗疟疾活性
疟疾,尤其是恶性疟原虫引起的疟疾,是致死性极高的传染性疾病,严重威胁人类健康。喹啉类抗疟疾药物是临床上应用最为广泛的抗疟疾药物之一,而喹诺酮可通过作用于疟原虫的红内期和肝期发挥抗疟疾功效。喹啉和喹诺酮二聚体可能具有双重作用机制,故可能具有更高的抗疟疾活性。4-氨基喹啉二聚体哌喹不仅抗恶性疟原虫和间日疟原虫引起的疟疾的活性不亚于氯喹,对耐氯喹恶性疟原虫引起的疟疾也有良好的疗效。目前,哌喹已被我国和印度尼西亚用于疟疾的防治。显然,喹啉和喹诺酮二聚体是抗疟新药优良的候选物。
SAR显示,4-氨基-7-氯喹啉结构片段可抑制β-血红素的形成,使药物在作用靶点累积,故4-氨基-7-氯喹啉结构单元对抗疟疾而言至关重要。无论用供电子基如氨基、甲氧基或吸电子基如硝基代替C-7位的氯,均会导致活性的大幅下降。因此,以不同连接子连接的4-氨基-7-氯二聚体引起了药物化学家的持续关注。
Vennerstrom等评价了烷基二胺(22,见图7)或杂环烷基二胺(23)连接的4-氨基-7-氯二聚体的体外(氯喹敏感型D6和耐氯喹型W2恶性疟原虫)和体内(伯氏疟原虫感染小鼠)活性,发现二聚体22 (IC50: 1.0~83 nmol/L)和23a-j (IC50: 1.2~89 nmol/L)对所测的氯喹敏感型D6和耐氯喹型W2恶性疟原虫均具有良好的活性,且对耐氯喹型W2恶性疟原虫的活性优于氯喹(IC50: 100 nmol/L)。SAR显示,含有哌嗪和带有醚键二胺连接子的二聚体在伯氏疟原虫感染小鼠模型中的体内活性优于相应的烷基二胺二聚体。体外活性最高的二聚体22m (Ro 48-6910, IC50: 1.0和1.4 nmol/L)具有良好的体内活性,其在给药剂量为160和320 mg/kg时的治愈率高达80%和100%。但Ro 48-6910与氯喹显示出体外交叉耐药性,且该二聚体在临床前研究中显示出光毒性,不具备进一步研究的价值。
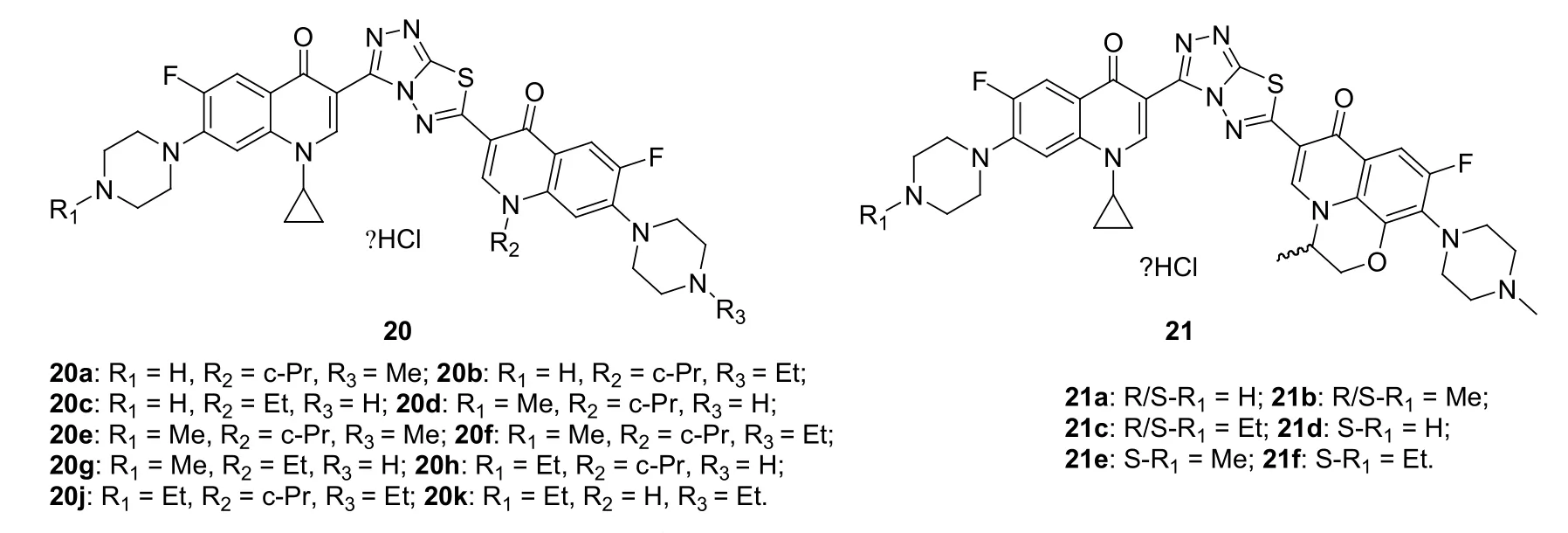
图6 氟喹诺酮二聚体20和21的化学结构
喹啉和喹诺酮二聚体由于体积比较大可能难以被转运蛋白外排,故这类二聚体可能能够克服疟原虫对氯喹的外排作用。喹啉二聚体24a (IC50: 98和89 nmol/L)和24b (IC50: 58和10 nmol/L)对耐氯喹型W2和K1恶性疟原虫的活性是氯喹(IC50: 280和154 nmol/L)的1.73~15.4倍,二者具有治疗耐药疟疾的潜力。对一系列含有直链或环胺基连接子的喹啉二聚体的体外抗恶性疟原虫活性和对哺乳细胞细胞毒性测试结果表明,与含有直链胺基连接子的二聚体相比,含有环胺连接子的二聚体尽管活性没有明显提高,但细胞毒性有所降低。活性最高的喹啉二聚体25a对耐氯喹型W2、FcB1R、D6和F32恶性疟原虫的IC50为64.6~170.1 nmol/L,与氯喹(IC50: 19~175 nmol/L)相当,且该二聚体对MRC-5和小鼠腹腔巨噬细胞无毒。二聚体25a的二茂铁衍生物25b对氯喹敏感型HB3和耐氯喹型Dd2恶性疟原虫也具有极高的活性,IC50为110和62 nmol/L,其抗氯喹敏感型HB3恶性疟原虫的活性优于氯喹 (IC50: 94 nmol/L)。大环多胺连接的7-氯喹啉二聚体25c (IC50: 4.1, 5.8和2.5 nmol/L)不仅无细胞毒性,而且对氯喹敏感型D6、耐氯喹型W2和耐甲氟喹型TM91C235恶性疟原虫的活性优于对照药氯喹(IC50: 8.2, 283.5和90.0 nmol/L)和甲氟喹(IC50: 19.9,7.2和55.8 nmol/L),具有治疗药敏型和耐药型疟疾的潜力。二聚体25c抑制β-血红素形成的IC50为1.1 μmol/L,是氯喹(IC50: 9.5 μmol/L)的8.6倍。在感染伯氏疟原虫的小鼠模型中,二聚体25c的体内活性与氯喹相当(半数有效量/ED50: ≤1.1 mg/kg; ED90: 5.6 mg/kg),且即使给药剂量为30 mg/kg仍未显示出体内毒性。以上结果表明,二聚体25c可作为候选物进一步研究。
脲连接的7-氯喹啉二聚体26可通过抑制血红素的形成抑制耐氯喹W2恶性疟原虫,其活性(IC50: 77 nmol/L)是氯喹(IC50: 244 nmol/L)的3倍。甾体连接的4-氨基-7-氯二聚体27对耐氯喹型W2和耐甲氟喹型TM91C235恶性疟原虫的活性与氯喹相当或更优,但对氯喹敏感型D6恶性疟原虫的活性弱于氯喹。研究表明,短链(n=0)二聚体27a (IC50: 119.64, 98.19和124.15 nmol/L)对所测氯喹敏感型D6、耐氯喹型W2和耐甲氟喹型TM91C235恶性疟原虫的活性优于长链(n=1)衍生物27b (IC50: 143.32, 108.31和194.74 nmol/L)。7-氯喹啉二聚体四氟硼酸盐28对氯喹敏感型Nigerian和耐氯喹型Fcba及FcM29恶性疟原虫的IC50为350~468 nmol/L,但弱于母药青蒿素(IC50: 4~8 nmol/L)和氯喹(IC50: 30~260 nmol/L)。
7-氯喹啉二聚体29 (IC50: 35.49~128.59 nmol/L)对D10和Dd2恶性疟原虫的活性优于其5-甲基衍生物(IC50: 80.22~1914.01 nmol/L),且对CHO细胞无毒。SAR显示,向R1位引入甲基对活性有利,且甲基二聚体29d (IC50: 37.38和71.13 nmol/L)对D10和Dd2恶性疟原虫的活性高于氯喹(IC50: 48.35和242.30 nmol/L)。二聚体29d对CHO 的CC50为7.8 μmol/L,选择性指数(SI)为211。进一步研究发现,这类二聚体也具有潜在的抗肿瘤活性,且活性最高的二聚体29a对TK10、UACC62和MCF7肿瘤细胞系的活性优于依托泊苷。
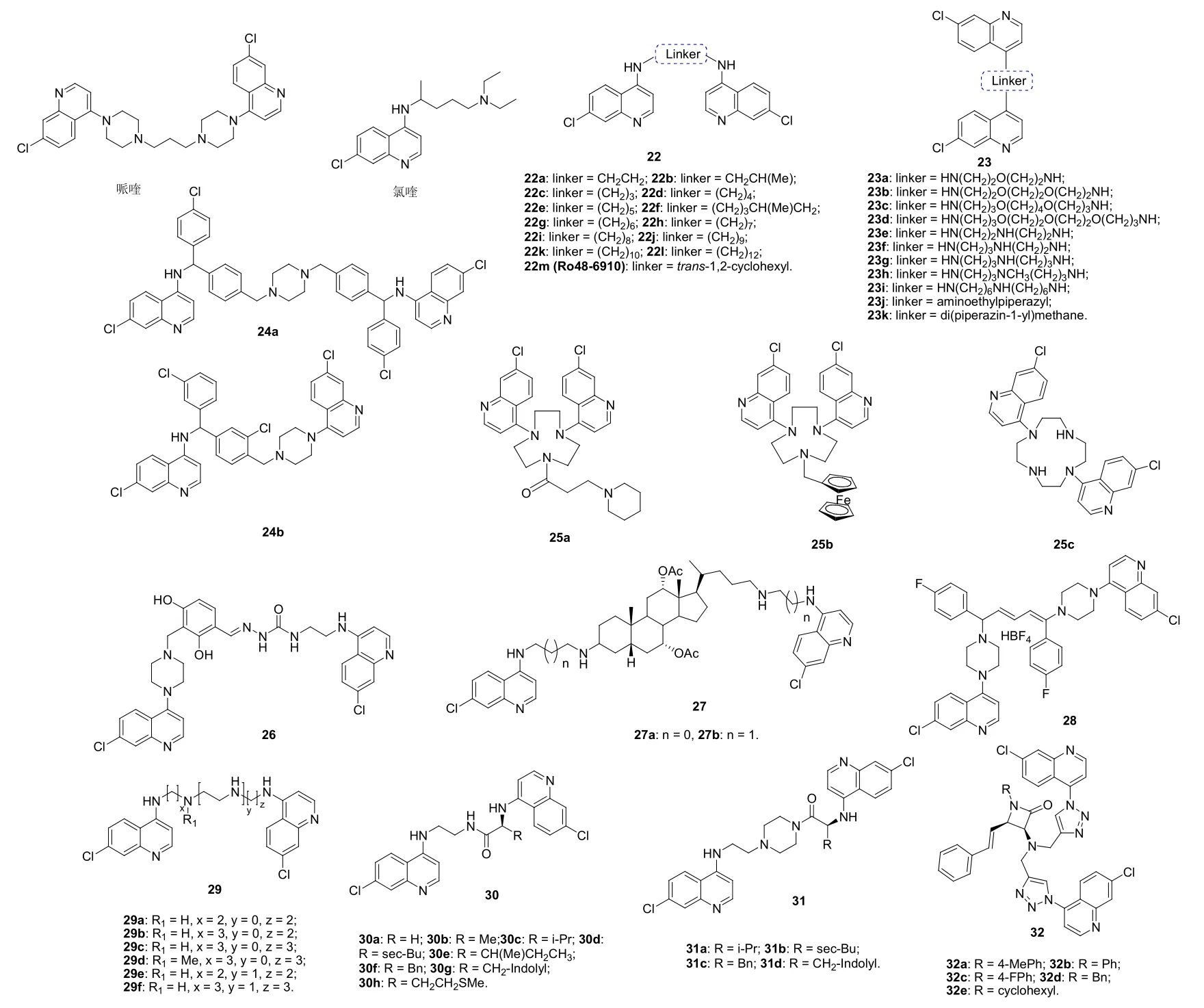
图7 7-氯喹啉二聚体22~32的化学结构
SAR研究显示,含有烷胺连接子的7-氯喹啉二聚体30 (IC50: 24~1430 nmol/L)的体外抗氯喹敏感型3D7和耐氯喹型K1恶性疟原虫的活性优于含有刚性哌嗪结构单元的二聚体31 (IC50: 355~2090 nmol/L)。侧链碳链的长度与二聚体的抗耐氯喹型K1恶性疟原虫活性息息相关,且短链如2个碳有利于活性,其中异丁基对活性最优。代表物30d对氯喹敏感型3D7和耐氯喹型K1恶性疟原虫的活性相当,IC50分别为24和26 nmol/L,对耐氯喹型K1恶性疟原虫的活性是氯喹(IC50: 255 nmol/L)的10倍。不仅如此,该二聚体30d的SI高达1011.71,值得进一步开发。
含有1,2,3-三氮唑和β-内酰胺结构片段的7-氯喹啉二聚体32仅显示出较弱的抗疟原虫活性,IC50在μmol/L水平,提示喹啉C-4位的仲胺对高活性而言至关重要。7-氯喹啉-吖啶衍生物具有弱到中等强度的体外抗NF54恶性疟原虫活性,对环状期进入裂殖期的寄生虫MIC为0.25~1 μg/mL。SAR显示,喹啉与吖啶之间的连接子对活性影响显著,且对/间苯二次甲基优于乙基二胺基和丙基二胺基。间苯二次甲基连接的二聚体33a (MIC: 0.25 μg/mL,见图8)的活性优于相应的对苯二次甲基连接的二聚体33b (MIC: 0.5 μg/mL),但均弱于氯喹(MIC: 0.125 μg/mL)。在伯氏疟原虫感染小鼠模型中,化合物33a可在给药d4(腹腔注射, 50 mg/kg)可完全消除小鼠体内的寄生虫血症,但小鼠的存活期不超过28d。
氯喹啉-喹啉化合物34a,b (IC50: 20.1和74.1 nmol/L)对耐氯喹型FcB1恶性疟原虫的活性优于氯喹(IC50:126.0 nmol/L)。其中,化合物34a (CC50: 16.4 μmol/L)对MRC-5细胞的细胞毒性低,SI为816,可作为先导物进一步优化。在伯氏疟原虫感染小鼠模型中,二聚体 35a尽管并未显示出任何活性,但其衍生物35b(ID50: 5.9 mg/kg; 14.9 mmol/kg)的活性与Ro 48-6910(ID50: 3.0 mg/kg; 8.3 mmol/kg)相当。
甲氟喹二聚体WR319691和WR319775对耐甲氟喹D6和耐多药恶性疟原虫C235 (对甲氟喹、氯喹和乙胺嘧啶耐药)具有良好的活性,IC90为22~80 ng/mL。WR319691和WR319775的甲基衍生物WR621613和WR621612对耐甲氟喹D6和耐多药恶性疟原虫C235的IC90: ≥329 ng/mL,提示甲基的引入对活性不利。
氯喹的耐药性主要由氯喹抗性转运体(PfCRT)突变导致,耐药性氯喹抗性转运体(PfCRTCQR)可通过介导寄生虫的消化液泡外排氯喹,且PfCRTCQR也可降低寄生虫对其它喹啉类抗疟药的敏感性。显然,PfCRTCQR是潜在的抗耐药疟疾药物的作用靶点。对一系列酯基、羰酰胺或酰胺连接的奎宁抑PfCRTCQR、体外抗恶性疟原虫和体内抗疟疾研究结果表明,酰胺36a和羰酰胺36b对PfCRTCQR具有良好的抑制作用,IC50为5.3和1.4 μmol/L,远优于奎宁、维拉帕米和沙奎那韦(IC50: 48, 30和13 μmol/L)。值得注意的是,二者并不会通过消化液泡被PfCRTCQR外排,而是在细胞器内累积。二者对β-血红素的抑制作用呈剂量依赖性,IC50分别为3.4和2.5 μmol/L,与奎宁(IC50: 3.9 μmol/L)相当。二者在血浆中的半衰期>90 h,提示稳定性良好。二聚体36a,b(IC50: 32.0~514.1 nmol/L)抑制氯喹敏感型HB3 (携带PfCRTCQS)、耐氯喹型Dd2 (携带PfCRTCQR)、FCB和P31 (携带PfCRTCQR)恶性疟原虫的IC50在纳摩尔级,尽管二者对氯喹敏感型HB3恶性疟原虫的活性弱于奎宁,但对所测所有耐药恶性疟原虫的活性均高于奎宁。在伯氏疟原虫感染小鼠模型中,二聚体36a(ED50: 38.2 mg/kg)的体内活性略优于奎宁(ED50: 48.7 mg/kg),但化合物36b在给药剂量为10和40 mg/kg时未显示出明显的抗疟疾活性。

图8 C-4位连接的喹啉二聚体33~38的化学结构
他克林二聚体37 (IC50: 50~6110 nmol/L)对氯喹敏感型3D7和耐氯喹型Dd2恶性疟原虫的活性与连接子息息相关,且长链(n≥5)连接子优于短链(n=1~4)连接子。进一步修饰所得的二聚体38 (IC50: 20~50 nmol/L)对氯喹敏感型3D7恶性疟原虫的活性与氯喹(IC50:20 nmol/L)相当,其中,代表物38f (IC50: 20 nmol/L)对J774.1的细胞毒性(CC50: 25 μmol/L)较低,SI为1,250。作用机制研究结果显示,二聚体38f (IC50: 5.2 μmol/L)可作用于寄生虫生长所必需的半胱氨酸蛋白酶falcipain-2,进而发挥抗疟疾功效。
噻吩[3,2-c]喹啉二聚体对氯喹敏感型3D7和耐氯喹型Dd2恶性疟原虫具有潜在的活性,IC50分别为210~2100 nmol/L和50~4100 nmol/L,弱于对照药氯喹(IC50: 21和210 nmol/L)。其中,二聚体39 (腹腔注射,见图9)在文氏疟原虫感染小鼠模型中的ED50为30 mg/kg。氯喹二聚体40对氯喹敏感型D10和耐氯喹型K1恶性疟原虫的IC50分别为43~454和17~25 nmol/L,耐药性指数(RI)≤0.5,说明这类二聚体具有治疗耐药疟疾的潜力。其中,二聚体40c (IC50: 43和17 nmol/L)对氯喹敏感型D10恶性疟原虫的活性与氯喹(IC50: 40和540 nmol/L)相当,对耐氯喹型K1恶性疟原虫的活性则是氯喹的31.7倍。
喹啉二聚体41、42和43对氯喹敏感型D10、耐氯喹型K1 (对氯喹耐药, 但对甲氟喹敏感)和K1mef (耐甲氟喹)恶性疟原虫的IC50为20~980 nmol/L,且活性顺序为42>43>41。含有4个碳连接子的二聚体活性最高,二聚体44也观察到了类似结果。代表物42d对耐药恶性疟原虫的活性优于对敏感恶性疟原虫,对氯喹敏感型D10、耐氯喹型K1和K1mef恶性疟原虫的IC50分别为50、20和20 nmol/L。在感染尼日利亚约氏疟原虫的小鼠模型中,二聚体42d (IC50: 5.2 mg/kg)的体内活性弱于氯喹(IC50: 2.5 mg/kg)。在给药剂量高于IC502倍时,二聚体42d显示出严重的毒性;当口服给药化合物42d剂量为25 mg/kg,尽管试验小鼠依然存活,但对体内尼日利亚约氏疟原虫的抑制率仅为13%。
Jain等评价了一系列8-氨基喹啉二聚体的体内外抗疟疾活性及对VERO细胞的细胞毒性,发现所有的二聚体均对VERO细胞无毒,且对氯喹敏感型D6和耐氯喹型W2恶性疟原虫的活性(IC50: 0.3~4.76 μg/mL)与首喹(IC50: 2.0和2.8 μg/mL)相当。其中,代表物45(IC50: 0.34和0.3 μg/mL)的体外抗氯喹敏感型D6和耐氯喹型W2恶性疟原虫活性是首喹的6和9倍。在伯氏疟原虫感染小鼠模型中,二聚体45可在给药剂量为10 mg/kg时治愈率达83.3%,给药剂量为25 mg/kg时完全抑制血内期疟原虫,而首喹即使在给药剂量为100 mg/kg时也未显示出任何活性。
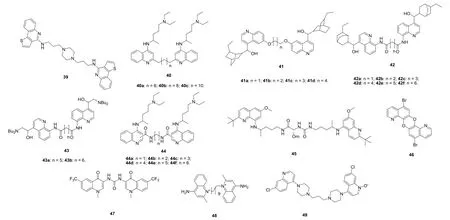
图9 喹啉二聚体39~49的化学结构
喹啉和喹诺酮二聚体46和47未显示出明显的体外抗疟原虫活性,而二聚体48具有良好的体内外活性。二哌喹的代谢产物49也具有潜在的抗疟原虫活性,值得进一步研究。
5 抗结核活性
结核病(TB)主要是由结核分枝杆菌(MTB)引起的致死性传染病,严重威胁人类健康。耐药TB (DRTB)尤其是耐多药TB (MDR-TB)的不断涌现和在世界范围内的广泛传播,已成为全球所必须共同面对的棘手问题。据世界卫生组织估计,仅2016年一年全球新增病例高达1040万,167万患者因此丧命。其中,新增耐利福平患者约60万,而其中的49万是更难治的MDR-TB患者。因此,亟需开发对药敏型和耐药型TB均有效的新型药物。
喹啉和喹诺酮衍生物具有良好的抗TB活性,其中的某些产品如贝达喹啉和环丙沙星已被用于治疗MDR-TB患者。分枝杆菌极度疏水的细胞膜是阻止抗TB药物到达作用靶点的天然屏障,故化合物的脂溶性对其抗TB活性至关重要。适当的提高化合物的脂溶性可能会提高其抗TB活性,而喹啉而喹诺酮二聚体的脂溶性高于相应的单核化合物,故喹啉而喹诺酮二聚体可能具有更高的抗TB活性,值得研究。
喹啉二聚体对MTB临床分离株(耐异烟肼、利福平、乙胺丁醇和环丙沙星)具有良好的活性,MIC为1.1~43.4 μmol/L。SAR显示,与无取代的二聚体50a-k(见图10)相比,在R2位含有氯原子的二聚体50l-q的活性更高。向苯环上引入卤素如氯和溴可提高活性,且卤素氯和溴在苯基的间位优于在对位,而供电子基的甲基和甲氧基对活性不利。二聚体50o,q(MIC: 1.1和2.2 μmol/L)对所测菌株的活性是环丙沙星、乙胺丁醇和吡嗪酰胺(MIC: 4.7, 7.6和50.77 μmol/L)的2.1~46.4倍,但弱于异烟肼和利福平(MIC: 0.4和0.1 μmol/L)。细胞毒性试验结果表明,二者小鼠成纤维细胞NIH 3T3的毒性极低,IC50分别为333和887 μmol/L。
对贝达喹啉衍生物51的体外抗MTB H37Rv和2株MDR-TB临床分离株的评价结果表明,除化合物51ce (MIC: >50 μmol/L)外,所有衍生物(MIC: 0.39~3.12 μmol/L)均具有潜在的活性。SAR表明,R1位为甲基对活性最优,而向苯环的间位(R2位)引入溴对活性不利。活性最高的化合物51a,g对MTB H37Rv和2株MDR-TB临床分离株的MIC分别为0.39和3.12 μmol/L,且对VERO和小鼠骨髓来源巨噬细胞无毒(CC50:>100 μmol/L)。化合物51a,g可分别降低90%和91%的小鼠骨髓巨噬细胞内的MTB菌落单位,与异烟肼和利福平(可降低98%的菌落单位)相当。在骨髓巨噬细胞内模型中也得到了类似的结果。在MTB H37Rv感染的小鼠模型中,二聚体51a的体内活性不亚于异烟肼和乙胺丁醇。当给药剂量为50 mg/kg时,所有小鼠在d29仍存活,但给药剂量增加到100 mg/kg时在d9有1只小鼠死亡。
腙连接的喹啉二聚体52 (MIC: 2.2~9.6 μmol/L)对MTB H37Rv具有中等强度的活性,但弱于异烟肼和利福平(MIC: 0.36和0.037 μmol/L),但这类化合物对VERO 细胞的毒性较高,SI在1左右。大多数2-氯喹啉二聚体53对MTB H37Rv无活性, MIC90>30 μg/mL。活性最高的化合物53a对MTB H37Rv和M. bovis BCG的MIC90为7.8和9.4 μg/mL,远弱于利福平(MIC:0.02和0.0173 μmol/L)。
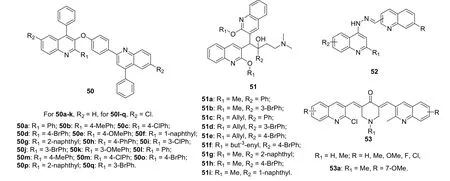
图10 喹啉二聚体50~53的化学结构
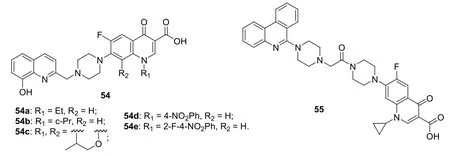
图11 喹啉-氟喹诺酮化合物54和55的化学结构
喹啉-氟喹诺酮(诺氟沙星、环丙沙星和氧氟沙星)衍生物54a-c(见图11)在浓度为6.25 μg/mL时对MTB H37Rv的抑制率分别为86%、98%和98%,活性优于N-1位芳基衍生物54d,e (6.25 μg/mL时对MTB H37Rv的抑制率为44%和47%)。菲啶-环丙沙星衍生物55 (MIC:12.5 μg/mL)对MTB H37Rv具有中等强度的活性,但弱于异烟肼和利福平(MIC: 0.36和0.02 μg/mL)。
6 其它生物活性

图12 喹啉二聚体56~58的化学结构
除以上生物活性外,喹啉和喹诺酮二聚体还具有其它活性。喹啉二聚体56a-f(见图12)对杜氏利什曼原虫前鞭毛体和胞内无鞭毛体具有良好的活性,IC50分别为2.0~13.5 μg/mL和2.1~10 μg/mL。SAR显示,向R1位引入甲基对活性不利,而向R2位引入氯或延长碳链长度对活性有利。其中,二聚体56a、56b和56f (IC50: 2.0~2.8 μg/mL)对杜氏利什曼原虫前鞭毛体和胞内无鞭毛体的活性与喷他脒(IC50: 2.1和2.8 μg/mL)相当,但弱于amphotericitin B (IC50: 0.16和0.1 μg/mL)。在内脏利什曼病BALB/c小鼠模型中,三者未显示出明显的毒性。其中,代表物56b在给药剂量为12.5和5.5 mg/kg时可降低95%和98.49%的脾组织和肝脏寄生虫,且在给药剂量>12.5 mg/kg对肾脏和肝脏无毒。二聚体57a-c对亚马逊利什曼原虫无鞭毛体和硕大利什曼原虫无鞭毛体无活性(IC50>32 μmol/L),但对婴儿利什曼原虫无鞭毛体和布氏锥虫无鞭毛体具有一定的活性,IC50为8~24 μmol/L。
喹啉二聚体58对秀丽隐杆线虫、多形螺旋线虫和阴道毛滴虫的活性与对照药相当或略弱于对照药。喹啉二聚体58对乙酰胆碱酯酶(AChE)的活性弱于丙硫咪唑,其在200 mg/kg时可降低31.2%的体内旋毛虫,但弱于甲苯达唑(在25 mg/kg时可降低84.2%的体内旋毛虫)。
6 结束语
喹啉和喹诺酮二聚体具有多种药理活性如抗菌、抗肿瘤、抗疟疾和抗结核活性,其中某些喹啉和喹诺酮二聚体已在临床上使用。因此,近年来喹啉和喹诺酮二聚体引起了药物化学家的广泛关注。近年来,药物化学家有针对性的设计、合成并评价了众多喹啉和喹诺酮二聚体的体内外生物活性,发现了若干有苗头的化合物。本文着重介绍了近年来该领域的最新研究进展,并归纳这类化合物的构-效关系,以期指导药物化学家更合理的设计此类二聚体。
