一种α,β不饱和羧酸亚胺酯的合成、表征与晶体结构
2019-10-11吴滨范喆王吉庆杨金明
吴滨,范喆,王吉庆,杨金明
(中南民族大学 药学院,武汉 430074)
CuAAC(Copper-Catalyzed Azide-Alkyne Cycload dition)反应,是指在Cu(I)催化剂存在的条件下,叠氮化物与末端炔烃通过分步反应发生1,3-环加成反应生成1,4-二取代-1,2,3-三氮唑的反应[1].该反应以其温和的反应条件、优异的收率和普适性引起了广泛的关注[2].1,4-二取代-1,2,3-三氮唑因其独特的化学结构也成了金属有机化学家们的一个热点研究领域.通常,在过渡金属铑(II)的催化作用下,1,4-二取代-1,2,3-三氮唑极易失去一分子氮生成α-亚胺基铑卡宾,从而有效地替代危险的重氮试剂.这种独特的反应模式使其成为一种非常稳定、安全的铑卡宾前体[3,4].三氮唑产生α-亚胺基铑卡宾后参与的反应非常丰富,多个课题组相继报道了该活性中间体与烯烃[5,6]、炔烃[7]、醛和亚胺[8]、α,β-不饱和醛[9]、联烯[10]、异氰酸酯和异硫氰酸酯[11]、以及1,3-二烯[12]的环加成反应,合成多种类型的杂环化合物.施敏小组报道了铑(II)催化官能团化的1,2,3-三氮唑类底物合成氮桥苯并二氧环庚烷衍生物[13],1,2-二氢异喹啉和1-茚酮类化合物[14],3-亚甲基-2,3-二氢苯并呋喃和3-亚甲基-2,3-二氢吲哚类化合物[15].几乎同时,利用相同的底物,仅仅改变反应条件,在体系中加入醇或者水作为亲核试剂,杨震课题组[16]报道了二氢异苯并呋喃和茚酮类化合物的合成.最近,李传莹小组[17]报道了铑(II)催化1,2,3-三氮唑与乙烯基醚化合物串联合成了系列哌啶衍生物.
从简单化合物合成具有结构多样性的复杂分子一直是有机合成领域的一个挑战问题,围绕click化学发展多组分串联反应为此问题提供了一个强有力的策略.近年发现,在一些炔烃化合物中,铜催化产生环加成中间过渡态后会脱去一分子氮生成烯酮亚胺的结构,烯酮亚胺中间体会进一步接受亲核试剂进攻,发生重排反应,得到α,β-不饱和亚胺化合物.Punniyamurthy小组[18]报道了一价铜催化炔醛,苯酚以及磺酰叠氮在温和条件下一步合成芳基甲基醚类香豆素衍生物,并提出了可能的机理.在一价铜的催化下,炔烃与磺酰叠氮发生[3+2]环加成生成中间体A,A失去一份子氮并再生一价铜催化剂后产生烯酮亚胺中间体B[19],氧上的孤对电子进攻中间体B,可能通过四元环两性离子过渡态C进行拟态周环的[1,3]-迁移重排[20,21],从而得到中间体D,再与苯氧基负离子发生1,4共轭加成反应和羟醛缩合反应最终得到目标化合物(图1).
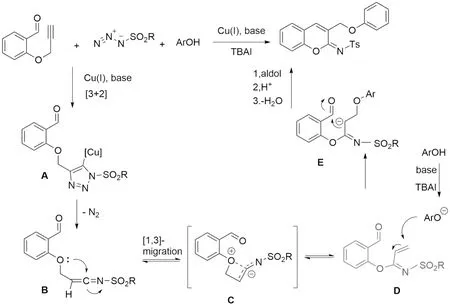
图1 Punniyamurthy小组提出的烯酮亚胺重排可能机制
为了进一步验证并完善该反应机理,设计通过水杨酸甲酯1a为原料合成底物2a,再通过CuAAC反应合成目标化合物3a.结果除了以21%的分离收率得到目标化合物3a之外,还意外得到了41%的主产物3b.核磁谱图及高分辨质谱对化合物3b的结构进行了初步表征解析,并进一步通过X-射线单晶衍射确定了化合物3b的结构(图2和3).

图2 化合物2a、3a以及3b的合成
1 实验部分
1.1 仪器与试剂
1H NMR在Bruker AM-600上测定,氘代试剂为Cambrige生产,TMS作为内标物,化学位移单位为ppm.HRMS在Agilent 6200 Q-TOF上测定.X-射线单晶衍射通过BRUKER D8 QUEST测定.水杨酸甲酯、炔丙基溴以及对甲苯磺酰叠氮等试剂从Alfa Aesar、韶远、安耐吉、阿拉丁、TCI、柏卡等公司购买.除甲苯进行了回流重蒸除水(CaH2),其他试剂使用前未经任何处理.
1.2 化合物2a的合成
称取水杨酸甲酯(30 mmol,3.9 mL)和碳酸钾(60 mmol,8.30 g)于50 mL圆底烧瓶中,加入20 mL N,N-二甲基甲酰胺(DMF)溶解,室温下搅拌,将炔丙基溴(36 mmol,3.1 mL)缓慢滴加进烧瓶中,搅拌过夜.加入200 mL水溶液淬灭反应,加入乙酸乙酯萃取3次,然后合并有机相,用饱和食盐水水洗有机相,再用无水硫酸钠干燥有机相,减压浓缩,通过柱层析进行粗分离纯化(石油醚/乙酸乙酯=10/1)得浅黄色油状液体2a6.1g.2a为已知化合物,1H NMR数据与文献报道[22]的一致.
1.3 化合物3a,3b的合成与3b的表征
在N2氛围下,将2a(5 mmol,957 mg)与CuTC(0.1 mmol,19 mg)加到50 mL圆底烧瓶中,加入干燥的甲苯(15 mL),随后边搅拌边缓慢滴加对甲苯磺酰叠氮(5 mmol,1.1 mL),室温下搅拌过夜.反应结束后,将溶液通过硅藻土过滤,乙酸乙酯冲洗,减压旋干浓缩,柱层析分离(石油醚/乙酸乙酯=5/1),得浅黄色固体442 mg3a,产率23%,白色固体741 mg3b,产率41%.
化合物3a.1H NMR (600 MHz,CDCl3)δ8.33 (s,1H),8.01 (d,J=8.4 Hz,2H),7.85 (dd,J=7.7,1.8 Hz,1H),7.47 (ddd,J=8.4,7.4,1.8 Hz,1H),7.41-7.36 (m,2H),7.08-7.02 (m,2H),5.29 (s,2H),3.91 (s,3H),2.45 (s,3H).13C NMR (151 MHz,CDCl3)δ166.34,157.56,147.56,144.44,133.87,132.97,132.08,130.58,128.87,122.90,121.55,120.71,114.16,63.23,52.22,21.98.
化合物3b.1H NMR (600 MHz,CDCl3)δ7.97 (dd,J=7.8,1.7 Hz,1H),7.56 (d,J=8.3 Hz,2H),7.53 (td,J=7.9,1.7 Hz,1H),7.35 (q,1H),7.30 (td,J=7.7,1.0 Hz,1H),7.17 (d,J=8.1 Hz,2H),7.10 (dd,J=8.1,0.9 Hz,1H),6.68 (dd,J=17.0,1.0 Hz,1H),6.16 (dd,J=10.9,1.0 Hz,1H),3.69 (s,3H),2.36 (s,3H).13C NMR (151 MHz,CDCl3)δ166.18,164.79,151.07,143.35,138.42,134.02,132.64,131.95,129.32,126.70,126.49,125.38,123.46,123.20,52.39,21.63.HRMS calcd for [C18H17NO5S] requires 359.0827,found 360.0892 [M++H]; 382.0712 [M++Na].
单晶培养:取50 mg3b溶解于少量二氯甲烷中,加入正己烷和正戊烷的混合溶剂后于室温下静置1 d,得到无色块状晶体.
1.4 晶体结构测定
配合物的单晶结构数据在BRUKER D8 QUEST,BrukerShelxTL软件包解析和优化该结构,多扫描方法(SADABS)对吸收效应进行数据校正,晶体结构用直接法求解,所有非氢原子使用全矩阵最小二乘法对F2进行各项异性修正,用理论加氢法对氢原子进行加和至理论位置.
2 结果与讨论
2.1 晶体结构鉴定和描述
化合物3b(C18H17NO5S)的晶体数据和有关数据收集及结构精修数据列于表1,相关键长和键角数据列于表2.晶体编号:JM1-29-1.化合物3b的晶体结构如图3所示,是一种无色块状晶体,属三斜晶系,P-1空间群,相对分子量M=359.38,近似尺寸为0.160 mm×0.337 mm×0.410 mm.X-ray晶体分析表明,晶胞参数a=7.9832(2)Å,b=8.3409(2)Å,c=14.5288(4)Å,α=81.1650(10)°,β=74.4740(10)°,γ=63.9960(10)°,V=837.03(4)Å3.

表1 化合物3b的晶体数据和结构精修数据
表2是烯酮亚胺重排区域的相关键长键角.因为晶体结构C1-C2-C3-C4-C5-C12-C11,包括O4-S1-O3区域是对甲苯磺酰基,C7-C16-C15-C14-C13-C8和O5-C9-O2-C10这部分对应水杨酸甲酯区域.根据晶体结构,C17-C18的碳碳双键与C6-N1的碳氮双键是处于反式共轭构型,因而能得到热力学稳定的3b.这为Punniyamurthy[6]小组提出的伪周环四元环状过渡态机理猜想提供了有力的证据,同时他们提出的机理中化合物D的画法是顺式共轭的构型,经过本实验的单晶验证,可以修正为反式共轭画法,这样更为严谨(图3).

图3 化合物3b的晶体结构图
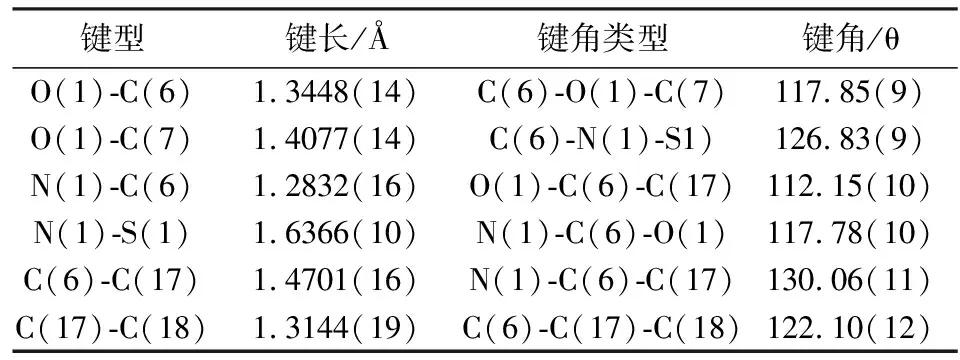
表2 配合物1的有关键角和键长数据
3 结语
使用CuAAC反应条件与设计的底物反应,意外合成出了化合物3b,通过1H NMR、HRMS以及X-射线单晶衍射证实了化合物3b的确切结构.首次报道了烯酮亚胺发生迁移重排反应后的产物的单晶结构,为烯酮亚胺的重排化学提供了有力的证据,并为烯酮亚胺作为有机反应活性中间体发展更多有机合成方法学提供了理论依据,基于烯酮亚胺为中间体的新型有机合成方法学的开发正在进一步探索中.
