亚胺自由基引发的1,5-氢原子转移反应研究进展
2022-02-19姜志洁
姜志洁
(陕西学前师范学院 化学化工学院,陕西 西安 710100)
亚胺自由基中氮原子以sp2杂化状态成键,亚胺自由基是σ自由基,其既有亲电性也有亲核性[1]。亚胺自由基参与的“C—N键形成反应”在合成含氮化合物中发挥着不可替代的作用。然而,缺少亚胺自由基清洁高效的产生方法和亚胺自由基高度活泼不易控制制约了亚胺自由基化学的发展。过渡金属催化和可见光氧化还原的蓬勃发展为亚胺自由基可控产生和引发的氢原子转移C(sp3)—H键官能团化反应提供了有效的解决方案,取得了一系列研究成果[2-4]。本文根据产生亚胺自由基的来源可分为肟的衍生物、亚胺类化合物、叠氮化合物三类,通过单电子还原或者氧化的方式可以产生亚胺自由基进行了总结。
1 肟的衍生物为亚胺自由基前体
肟的衍生物由于分子内存在较弱的N—O键(BDE≈50 kcal/mol),O-酰基肟在有机合成中得到广泛应用[5]。除了经典的Beckmann和Neber重排外,O-酰基肟可以通过氧化加成转化为有机金属物种[6],或者单电子转移生成亚胺自由基[7]。肟和酰氯经一步简单酯化反应制备肟酯,其经单电子还原断裂N—O键生成亚胺自由基。肟衍生羧酸化合物可以由酮与氨氧基羧酸制备,其经单电子氧化裂解N—O键生成亚胺自由基。Forrester[8]、Zard[9]等课题组在亚胺自由基参与的反应方面开展了一系列开创性工作。近年来随着可见光催化和金属催化的蓬勃发展,亚胺引发的1,5-HAT反应得到了深入的研究。
1.1 光催化亚胺引发的1,5-HAT反应
近十年来,光化学反应具有反应条件温和、官能团容忍性高、反应可控等优点在有机合成领域展现出重要应用价值[10]。过渡金属如铱、钌等与多吡啶类配体的复合物、有机染料分子、异质半导体等将光能可控的转化为化学能产生活性自由基中间体引发后续反应进行。
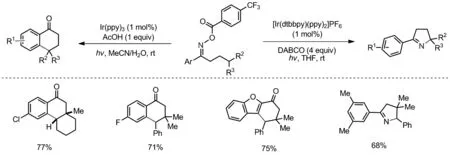
2017年,Nevado课题组报道了以Ir(ppy)3为催化剂经单电子还原肟酯,生成亚胺自由基引发1,5-HAT,合成了一系列苯并环己酮衍生物(Scheme 1)[11]。酸性条件下质子化的亚胺自由基亲电性增强,1,5-HAT过程能垒降低,加速反应进行。碱性条件下,选用[Ir(dtbbpy)(ppy)2]PF6为光催化剂且亚胺自由基HAT位点为苄基时,可实现分子内C(sp3)—N键构建,合成二氢吡咯化合物。该反应避免了使用Forrester报道中需要强酸高温体系,具有反应条件温和官能团兼容性好等优点。
Scheme 1
Scheme 2
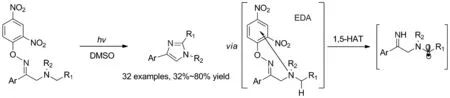
肟醚在无光催化剂条件下同样可以产生亚胺自由基,并参与分子内氢原子转移反应。2017年,付华课题组通过光诱导电子给体受体(electron donor-acceptor,EDA)复合物引发的电子转移机制,合成了天然产物和药物分子中常见的咪唑类化合物(Scheme 2)[12]。在白光灯照射下,肟醚底物中富电子的三级胺与缺电子的芳环发生单电子转移经历自由基离子对裂解N—O键生成亚胺自由基引发后续反应进行。该反应体系简单,不再需要昂贵的铱催化剂、酸碱添加剂,具有广泛的应用前景。
2017年,吴劼课题组同样利用分子间EDA机制,在无催化剂和添加剂条件下实现了环状磺酰亚胺的合成(Scheme 3)[13]。紫外-可见吸收光谱证实肟醚底物与DABSO形成EDA复合物。反应条件温和且具有较好的官能团兼容性和底物普适性。

Scheme 3

Scheme 4
Scheme 5
亚胺自由基引发的氢原子转移除了可通过分子内环化构建C(sp3)—C、C(sp3)—X键,还适用于分子间C(sp3)—C、C(sp3)—X键的构建。
2018年,段新华课题组在蓝光条件下,以Ir(ppy)3为光催化剂,烯烃为烷基化试剂,醇和水为亲核试剂,生成了γ-羟烷基化的酮。其反应机理为单电子还原产生的亚胺自由基,首先发生1,5-HAT生成烷基自由基,选择性加成到C=C双键末端后生成的苄基自由基后再氧化成正离子,在H2O的亲核进攻下构筑C—O键,最终实现了酮的γ-羟烷基化(Scheme 4a)[14]。这是首例由肟酯类底物为亚胺自由基前体实现的分子间C(sp3)—C(sp3)键构建反应。通过将乙腈溶剂替换为二甲基亚砜,且体系加入当量TsOH的条件下,通过消除一分子水,得到Heck类型的产物(Scheme 4b)[15]。除了芳基烯烃,香豆素和雌酮衍生的烯烃都能在该条件下顺利与肟酯反应,可实现多种γ-烯基化酮化合物的合成。同期,俞寿云课题组利用相同的条件以肟酯和烯基硼酸作为底物,实现了经亚胺自由基1,5-HAT后合成γ-烯基化酮化合物[16]。
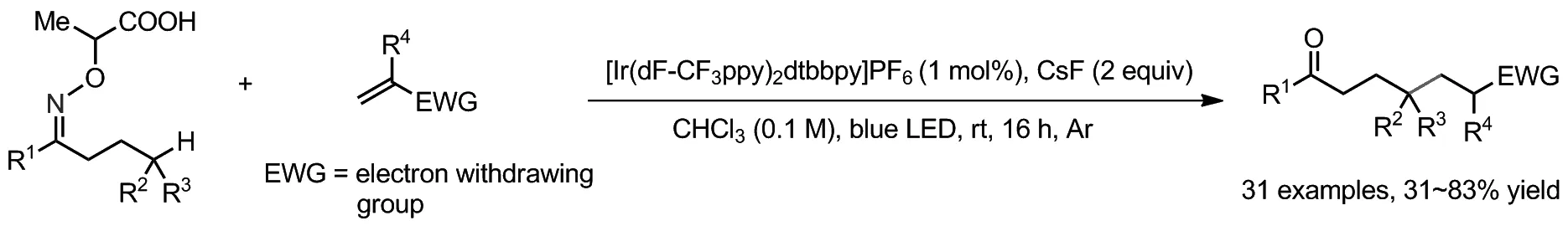
2018年,Studer课题组以链状肟衍生羧酸为亚胺自由基前体,通过Michael受体捕获亚胺自由基1,5-HAT后生成的烷基自由基,实现了烷基酮化合物γ-C(sp3)—H键烷基化反应(Scheme 5)[17]。该反应也实现了直接由C(sp3)—H键直接构建分子间C(sp3)—C(sp3)键反应,但存在较大局限性:肟衍生羧酸迁移位点局限在三级和苄基C(sp3)—H键;Michael受体也局限于α-芳基丙烯酸酯和α,β-不饱和酮类化合物。
Scheme 6
Scheme 7
Scheme 8

Scheme 9
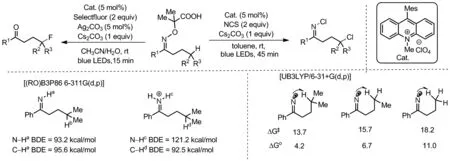
2018年,Leonori课题组利用以吖啶盐为光催化剂,Ag2CO3作为助催化剂,Selectfluor为氟源高效合成了γ-氟代酮和γ-氯-N-氯代亚胺(Scheme 6)[18]。详细的理论计算解释了肟酸底物反应位点局限在三级C(sp3)—H键这一现象。1,5-HAT位点由三级、二级到一级C—H时,反应过渡态吉布斯自由能变逐渐递增,反应速率降低。
1.2 过渡金属催化亚胺引发的1,5-HAT反应
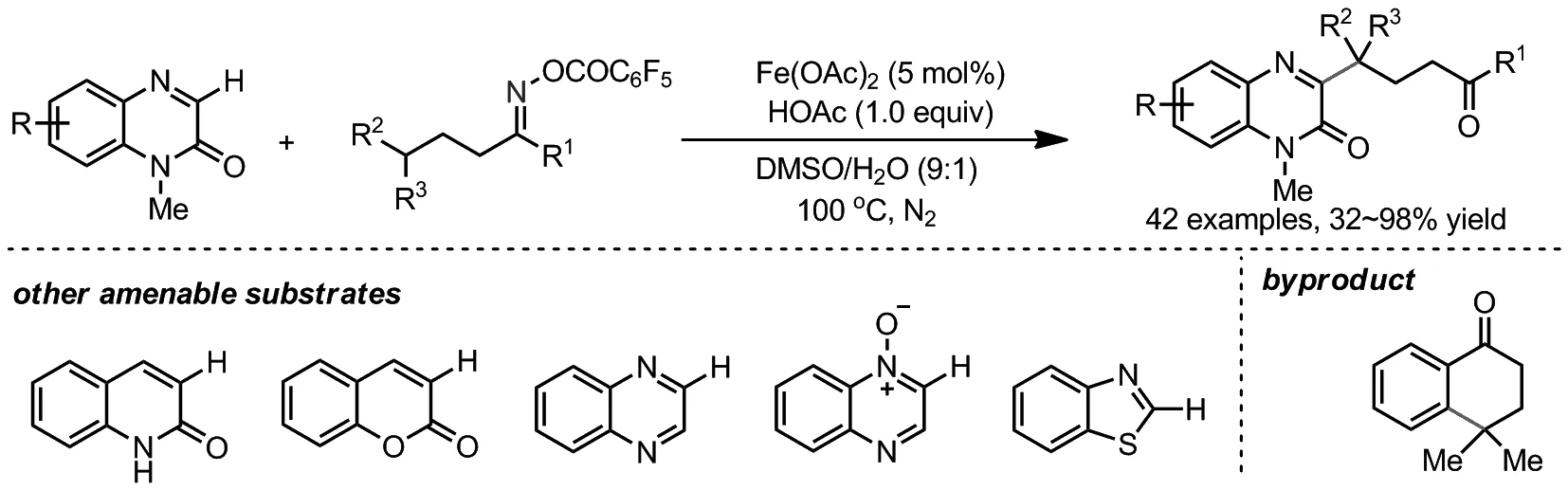
2019年,郭丽娜课题组报道了Fe(II)催化下,以肟酯和杂环化合物为底物,实现了烷基酮的远程C(sp3)—H键的杂芳基化反应(Scheme 7)[19]。除了喹喔啉酮外,喹啉酮、香豆素、喹喔啉、喹啉氮氧化物以及苯并噻唑都能顺利参与反应,得到一系列γ-杂芳基化的烷基酮。这一策略为实现芳杂环的酮烷基化提供了一条有效的途径。
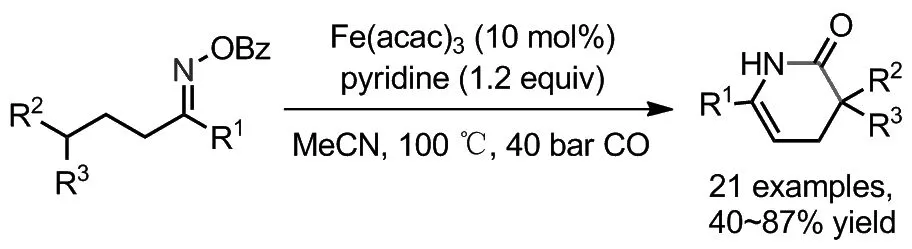
2019年,吴小锋课题组报道了Fe(acac)3催化肟酯经单电子还原生成亚胺自由基,经1,5-HAT生成的烷基自由基被CO顺利捕获,最后经分子内环化以高产率和优异的化学选择性地构建了各种六元内酰胺化合物(Scheme 8)[20]。这一工作扩展了由亚胺自由基氢原子转移后生成三级自由基的羰基化反应。
2019年,祝介平课题组报道了以Fe(acac)3为催化剂,TMSN3为氮源,实现肟酯γ-C(sp3)—H的叠氮化合成了γ-叠氮酮(Scheme 9)[21]。类似于郭丽娜课题组报道1 eq.HOAc为添加剂加速了1,5-HAT速度有利于提高反应收率。
Scheme 10
Scheme 11

Scheme 12
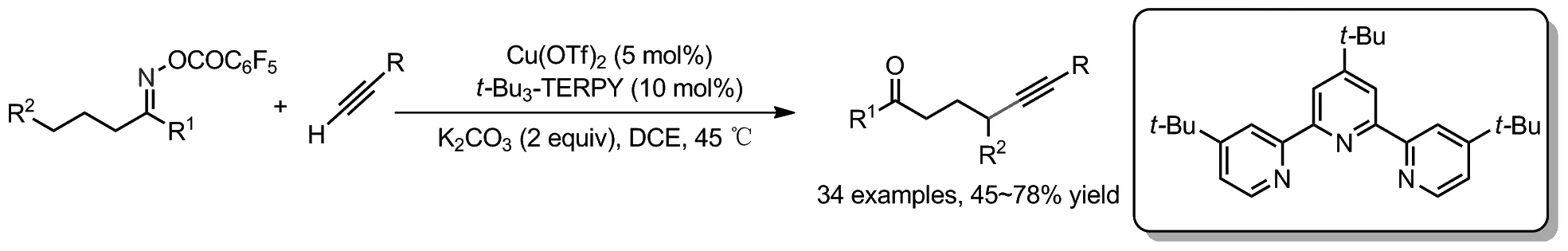
2020年,祝介平课题组延续前期工作报告了以Cu(OTf)2为催化剂,t-Bu3-TERPY为配体,肟酯γ-C(sp3)—H与末端炔烃的炔基偶联反应,合成了各种γ-炔基酮化合物(Scheme 10)[22]。
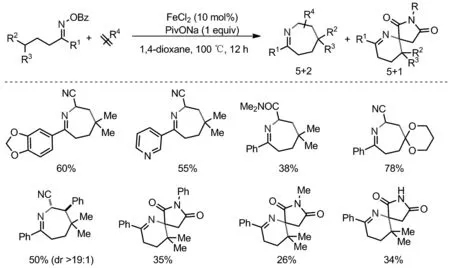
在上述亚胺自由基氢原子转移后构建分子间化学键反应过程中,亚胺最后都水解为羰基因此局限于烷基酮化合物的合成。2020年,陈应春课题组发展了一种FeCl2催化以肟酯为五原子结构单元,利用亚胺自由基引发的1,5-HAT/(5+2)或(5+1)与各种缺电子烯烃串联反应,合成了许多天然产物的核心结构或者生物活性分子的氮杂环庚烷化合物和螺-琥珀酰亚胺-四氢吡啶衍生物(Scheme 11)[23]。通过引入缺电子烯烃作为C2或C1单元,实现了亚胺1,5—HAT后分子间C(sp3)—C(sp3)键和C(sp3)—N键的构建。该反应具有底物范围广,官能团兼容性好的特点。
2 亚胺类化合物为亚胺自由基前体
2011年,Chiba课题组报道了Cu(II)催化亚胺自由基诱导的苄基C(sp3)—H键氧化反应合成1,2-二酰基苯类化合物(Scheme 12)[24]。格氏试剂和苯腈加成生成芳基亚胺。氧气氛围下,Cu(II)氧化亚胺得到自由基,1,5-HAT后生成苄基自由基并且成功捕获氧气生成过氧自由基通过苄位氧化和亚胺基水解得到1,4-二羰基化合物。通过底物结构设计,该课题组在类似条件下以脒作为底物,通过自由基的氢迁移策略实现了二氢恶唑的合成[25]。
2018年,陈弓、何刚课题组报道了加热条件下,NIS催化亚胺酸酯底物合成β-胺基醇和2-芳基噁唑啉的反应(Scheme 13)[26]。在化学当量的NIS条件下原位生成N—I键,通过N—I键均裂生成亚胺自由基。该反应具有非常广的底物范围,且一级、二级、三级β-C(sp3)—H键都可高效胺化。当R1为芳基时,生成的2-芳基噁唑啉较稳定,可经柱层析分离纯化;当R1为三氯乙酰基时,产物不稳定经酸解得到胺的盐酸盐。这一研究为芳基噁唑啉类化合物的合成提供了便捷的方法。

Scheme 13
Scheme 14
Scheme 15
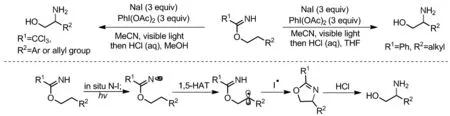
亚胺酸酯在光照条件下,同样可以产生亚胺自由基参与HAT反应。2017年,Nagib课题组报道了光诱导以亚胺酸酯为底物合成了β-胺基醇(Scheme 14)[27]。在NaI和PhI(OAc)2原位生成I3-催化磺酰胺分子内胺化策略可高收率得到C(sp3)—H键胺化产物。反应机理如下:光照条件下,均裂原位生成的亚胺酸酯N—I键生成亚胺自由基。随后经1,5-HAT生成β-位碳自由基,该自由基与碘自由基结合,再经分子内亲核取代生成噁唑啉,酸解生成β-胺基醇。以三氯乙酰亚胺酸酯为底物可实现苄基和烯丙基C(sp3)—H键的胺化;以苯酰亚胺酸酯为底物则实现键能较高的二级和一级C(sp3)—H键胺化。后续该课题组利用亚胺酸酯底物经1,5-HAT,实现了β-双卤代醇、噁唑和咪唑的合成[28-29]。

Scheme 16
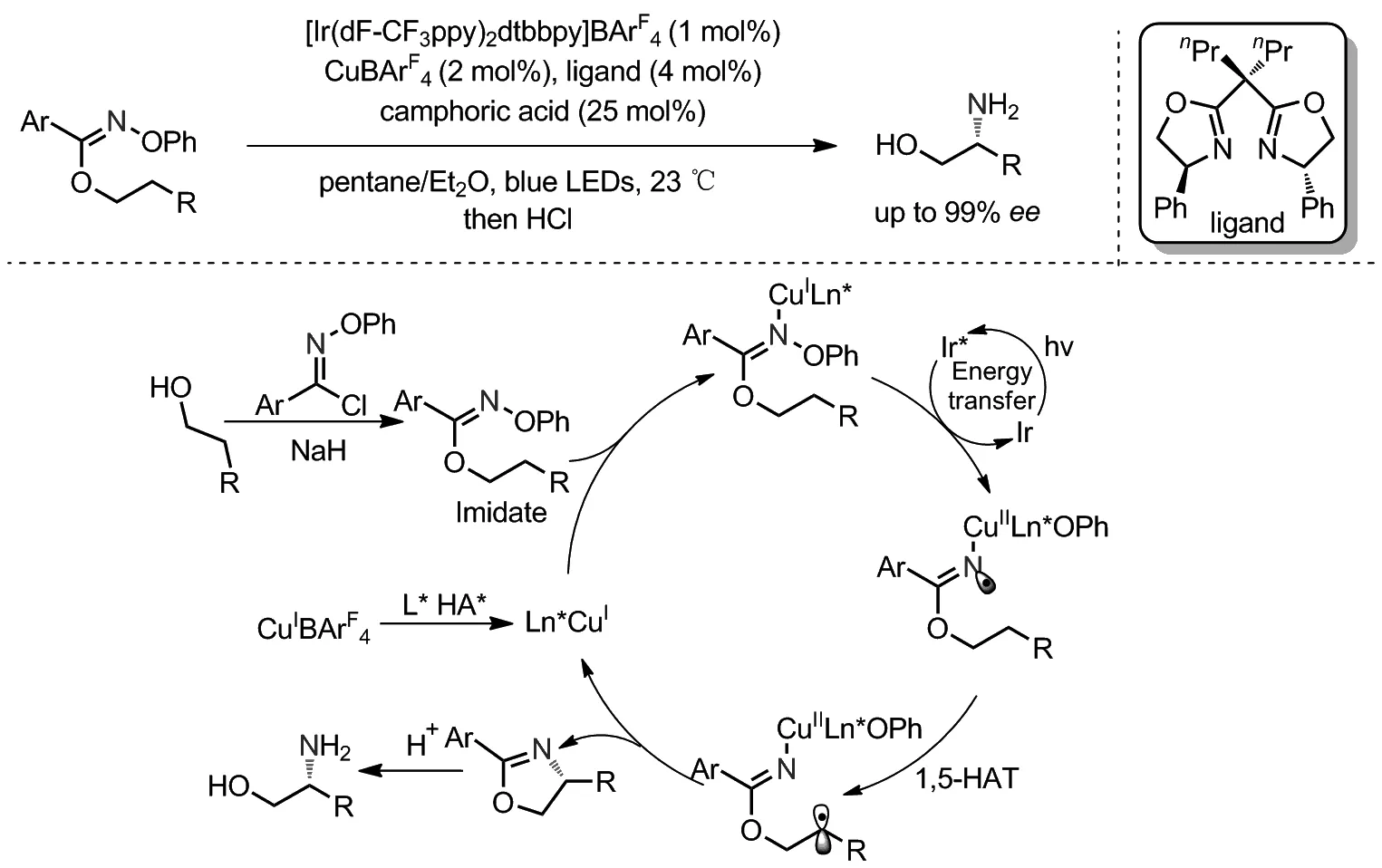
2020年,Nagib课题组通过光诱导铜催化不对称自由基碳氢键胺化实现了手性β-胺基醇的合成(Scheme 15)[30]。手性双噁唑啉配体既控制反应选择性又影响反应效率,阴离子B(ArF)4可以有效增加立体选择性控制。此外,樟脑酸作为共催化剂增加反应速率。该课题组基于理论计算、光淬灭实验、同位素标记实验、竞争实验、去对称化实验以及自由基Clock实验提出如下反应机理:醇与亚胺酰氯反应生成亚胺酸酯底物,与手性铜催化剂配位生成中间体。在可见光激发下,光催化剂通过能量转移活化铜络合物,通过氧化加成生成亚胺酸酯自由基,其经过1,5-HAT得到β烷基自由基,中间体最后经分子内胺化伴随近一步立体控制得到手性噁唑啉。最后,在酸性条件水解生成手性β-氨基醇。这是首例利用亚胺自由基1,5-HAT后实现的高对应选择性反应。
叠氮化合物生成亚胺自由基经分子内环化合成各种氮杂环化合物已得到广泛研究,但叠氮化合物作为亚胺自由基前体引发的HAT反应鲜有报道[31]。2017年,Nevado课题组报道了以烷基羧酸和烯基叠氮为底物,以Ag2CO3为催化剂,K2S2O8为氧化剂,高立体选择性合成了一系列苯并环己酮化合物(Scheme 16)[32]。该反应经历了氧化脱羧、自由基加成、脱氮气、亚胺自由基引发1,5-HAT、分子内环化等串联过程实现了目标产物的合成。该策略可高效、简洁地合成天然产物和药物分子,这也为复杂药物分子后期引入羰基提供了思路。机理验证实验和理论计算表明,1,5-HAT过程是反应的决速步骤。
亚胺自由基引发的氢原子转移反应是合成烷基酮以及氮杂环化合物的重要方法。目前,可控产生亚胺自由基及其引发的1,5-氢原子转移构建分子内C(sp3)—C键、C(sp3)—N键以及构建分子间C(sp3)—C键、C(sp3)—N键、C(sp3)—X键取得了系列研究进展,但仍然存在巨大挑战。例如,亚胺基自由基引发的1,5-HAT实现C(sp3)—H键官能化的多组分反应值得更多关注;探索新的反应模式,尤其通过将光氧化还原催化与其他成熟的催化技术(如过渡金属催化、有机催化和生物催化)相结合也是重点研究方向;鉴于亚胺自由基的高活性其参与的立体选择性反应研究较少,利用不对称自由基接力策略实现亚胺自由基引发的1,5-HAT后立体选择性C(sp3)—H键官能团化反应也是未来的发展方向之一。
