从作物基因组分析到整合组学知识库建设
2019-09-24梁承志
梁承志
从作物基因组分析到整合组学知识库建设
梁承志1,2
1. 中国科学院遗传与发育生物学研究所,植物基因组学国家重点实验室,种子创新研究院,北京 100101 2. 中国科学院大学,北京 100049
高通量技术的广泛应用使得各类组学数据的产出速度越来越快,由此产生的海量数据蕴藏着大量的基因组变异和相关功能信息。如何对这些数据进行深度整合和利用将会是一个长期而艰巨的任务,这需要具备高效的数据存储、分析和挖掘的能力。在过去几年中,本课题组通过与所内外课题组的合作,在多个植物的基因组的组装、注释、比较基因组和群体基因组分析等方面进行了探索,同时也将大量的水稻种质信息和组学数据进行了整合,存储于结构化数据库中并开发了一些相应的网络查询展示和数据挖掘工具。本文对相关的研究成果及其进展进行了概括性介绍,并展望了下一步的目标:构建一个用于支持作物功能基因组学和分子设计育种研究的整合组学知识库。
基因组分析;数据库;组学大数据;分子设计育种;作物;组学知识库
近年来,随着高通量组学技术的迅猛发展,各类组学数据的产出成本越来越低,产出速度也越来越快。这些新技术和海量数据已为动植物功能基因组、人类健康及遗传育种方面的研究带来了前所未有的机遇和挑战。一方面,基因组技术的广泛应用极大提高了这些领域的研究效率;同时,通过多学科知识、多领域技术和各类组学信息的交叉融汇,这些领域的前沿应用技术也在迅猛发展。如在作物育种方面,利用功能基因组的研究成果和高通量基因型表型测量技术,结合对已知优异等位基因聚合[1]和全基因组选择[2]等技术的品种智能设计[3~7]将是未来育种的必然发展方向,预期能够突破传统育种的瓶颈[8],大大提高育种选择精度,缩短育种周期。另一方面,虽然这些海量组学数据中蕴藏着大量基因组变异及相关的功能信息,但由于生物数据的高复杂性,人们对这些数据的深度整合、解析及重复利用将是一个长期艰巨的任务,特别是需要具备高效的数据存储管理,自动化分析和挖掘的能力。
中国科学院遗传与发育生物学研究所长期致力于研究重要农作物的分子遗传和分子育种中的重大科学和关键技术问题。为解决对海量组学数据的深度加工和广泛应用,并基于现有及未来科研项目的需求,研究所积极参与北京生命科学大型仪器区域中心相关平台的建设工作,在过去几年购置了“农作物分子育种数据分析系统”和“农作物大数据分析整合计算系统”。两套系统整体聚合计算能力超过50万亿次/秒,CPU核心数达1000多个,裸存储总量大于3 PB (图1)。在此基础上,2019年研究所成立了“生物大数据分析平台”,以加强对组学数据的管理和对上述计算存储资源的高效利用。平台的使命是服务于研究所的生物数据分析需求,通过提供培训服务和项目合作,致力于提高研究所的生物信息技术水平。同时,平台承担的一个重要任务是对研究所“一三五”战略规划建设、中国科学院战略性先导科技专项、种子创新研究院建设等提供组学大数据相关的技术支撑。在过去几年中,通过与所内外课题组的合作,本课题组利用上述计算存储系统,在多个植物的基因组分析方面做了一些较为深入的探索,同时也已将大量的水稻组学数据整合到了结构化数据库中并开发了一些相应的查询展示和挖掘工具。本文对本课题组取得的相关研究成果及其进展进行了概括性总结,并对未来构建一个主要作物整合组学知识库的发展目标进行了展望。
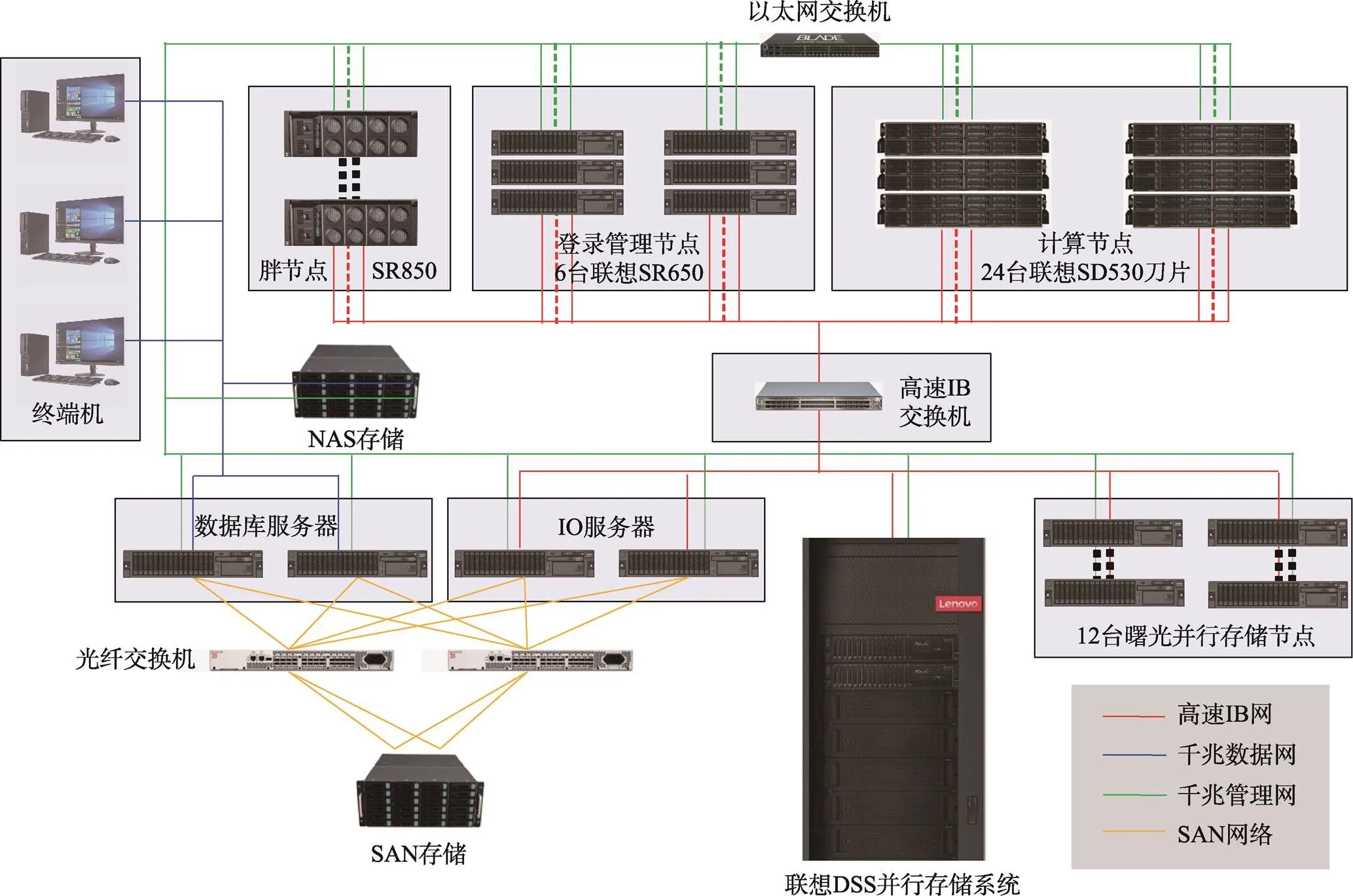
图1 中国科学院遗传与发育生物学研究所高性能计算集群及数据存储主体架构示意图
集群计算节点结合DSS并行存储用于大规模数据分析。SAN存储用于结构化数据库中数据存储,为网络服务器提供数据服务(有部分服务器未显示)。
1 研究成果及进展
1.1 基因组组装和注释
基因组组装和注释是基因组学研究的两个重要基础。本课题组通过结合全基因组PacBio (Pacific Biosciences)单分子测序、fosmid大片段DNA测序、遗传图谱和BioNano (BioNano Genomics)光学图谱等技术,利用已有的组装软件及自己开发的软件,构建了一个植物中最高质量的参考基因组水稻(ssp)蜀恢498序列[9]。利用类似的方法,通过所内外课题组合作解析了苦荞()[10]和二倍体小麦乌拉尔图()[11]的基因组。另外,本课题组还合作完成了栽培金鱼草()的基因组组装[12]。在此基础上,开发了一个用于基因组复杂区域局部组装的新软件HERA[13],能够在不借助于大片段DNA文库的情况下,组装出超长连续的基因组序列。利用HERA软件改进了多个已发表基因组,其中玉米和苦荞的基因组序列质量都已得到了巨大提高,包括序列的连续性提高了近50倍,多处错误得到了修正。目前本课题组已经利用HERA软件跟所内外多个课题组合作完成了数十个水稻、大豆、小麦、玉米和高粱等作物的高质量基因组序列(未发表数据)。对于任一纯合的二倍体水稻基因组,在HERA的帮助下都能得到一个与日本晴参考基因组质量相当的组装序列。对于杂合度高的基因组如野生稻,利用HERA提高组装序列的长度,再结合Hi-C数据辅助分离单倍体基因组也获得了很好的结果。
在基因注释方面,本课题组通过利用RNA-seq数据提高对基因的覆盖度,改进了Gramene-Pipeline注释流程[14],结合其他证据,注释了水稻[9]、苦荞[10]、小麦[11]和棉花(未发表数据)等多个基因组的高质量基因集。通过比较发现,这些基于证据的基因集比已发表的基因集质量在很多方面有较大的提高[9]。
1.2 比较基因组和群体基因组分析
比较基因组分析可用于研究基因组中的结构变异、基因组和基因家族的进化及保守基因的功能。本课题组通过对水稻、小麦、苦荞和金鱼草等与其近源物种或模式植物进行比较分析研究了各个物种中的全基因组复制事件、基因组之间的变异和进化模式,以及重要基因家族的进化对物种进化的影响。例如,通过对苦荞的比较基因组分析,发现了芦丁代谢途径上的所有9个酶的编码基因家族,包括两个以前未被完整克隆的酶的编码基因[10]。同时,发现在苦荞中全基因组复制事件中导致大量转运蛋白家族及转录因子家族的扩增对于苦荞耐铝、耐逆境等能力的提高起着很重要的作用[10]。在对小麦A基因组的研究中,发现与水稻基因组相比,小麦基因组进化的速度明显加快,在共线区域丢失的基因变多,产生的新基因的数目也变多,这可能是由转座子快速扩增引起的小麦基因组扩张导致的[11]。此外,利用拟南芥、水稻、玉米和大豆的大规模RNA-seq数据构建了基因调控和共表达网络并进行了比较分析[15],找到了一些物种之间保守的基因模块及跟重要农艺性状相关联的候选基因。这些模块对于研究基因的生物学功能提供了很重要的线索。
群体基因组研究可用于基因定位及物种驯化和进化分析[16]。在中国科学院战略性先导科技专项(A)“分子模块育种创新体系”资助下,本课题组与所内外多个课题组合作,对1000多份中国水稻主栽品种进行了重测序并测定了多环境下的多个表型数据(数据尚未发表,但可通过www.mbkbase.org查询)。通过对基因型信息的分析发现,中国的水稻品系在东北和南方粳稻、籼稻杂交稻亲本之间的分化非常明显,具有明显的群体结构。利用全基因组关联分析,发现了多个重要农艺性状的新关联位点。结合传统的群体基因组分析方法及本课题组自己发展的新方法,发现了一些各个亚群体之间的渗入片段,与目前的水稻育种目标(如提高品质和抗病性)具有很高的关联性(未发表数据)。此外,还分析了这些水稻主栽品种中对于已知优良等位基因的利用情况。这些结果为中国水稻优良基因的挖掘和进一步育种改良提供了线索。
1.3 数据库建设
进行上述基因组研究的一个重要目的是利用相关的分析技术和组学数据来构建一个整合多组学的数据库,以支持作物功能基因组学和分子设计育种研究。其中高质量的参考基因组及基因注释为种内不同个体的比较分析提供了全面的基础,而比较基因组及比较转录组研究可用于分析和关联重要基因的功能。对于群体基因组结合表型的关联分析为功能基因组研究到育种应用之间架起了一个信息和知识传递的桥梁。目前本课题组已初步完成了一个用于整合、结构化存储、查询、可视化展示、挖掘多组学数据的知识库MBKbase(www.mbkbase.org)中水稻子库的建设。水稻数据包括种质信息、表型、基于两个水稻参考基因组日本晴和蜀恢498的遗传变异、种质系谱树、种质基因组片段的亚群体祖先来源、已知和未知基因的等位变异及与表型信息的关联、功能基因的表达信息等。目前库中已收集了多个水稻群体的基因型和表型信息。尽管数据的系统性及软件的功能都尚有待于加强,这个知识库为基因型和表型数据的整合及深度高效利用已奠定了一个良好的基础。
2 作物多组学大数据整合知识库建设展望
当前生命科学研究涉及到越来越多的多学科交叉及多技术和多数据类型的整合,这不仅要求深入分析多种类型数据之间的关联,而且对于大数据整合分析工具的能力要求也越来越高。因此,建设一个能够整合大规模作物群体的多组学信息、提供大数据深度挖掘能力的知识库(图2)已是一个重要且急迫的任务。此知识库以整合的多组学信息为基础,特别是围绕作物大规模群体的基因型和表型组的关联并结合已知功能基因信息(图3),结合能够从基因型预测表型的模型和软件工具,对复杂性状进行分解预测。由于环境对作物的表型往往具有很大的影响,同一种基因型的材料在不同环境中的生长发育经常有很大差别,因此还需要整合环境信息以构建地区或环境特异的智能模型。在未来几年中,除了继续完善水稻子库,增加更多的水稻群体基因型和表型组信息外,本课题组将与植物染色体工程国家重点实验室多个课题组合作,充分利用其前期积累的数据,包括基因型、功能基因或遗传位点信息、基因表达数据、田间表型、代谢组数据等[17,18](及未发表数据),以期构建大豆、小麦等作物的整合多组学知识子库。
在分子育种技术方面,全基因组选择(genomic selection, GS)技术利用基因型来预测育种值,近年来已在国外大型商业育种项目中得到了广泛应用,大大减少了多环境测试的品系数目,在一些作物育种实例中减少了一半以上的传统谱系育种时间并增加了产量[19~21]。不过,目前GS技术在育种中的应用尚有很多的局限需要解决[19,20]。例如,GS的基因组预测模型有很多种,包括是否线性、有无参数、贝叶斯、机器学习等多种类型。GS模型的预测精度受到被选择性状的遗传结构、遗传力和基因的效应,训练群体的结构、大小和遗传多样性,测试群体与训练群体之间亲缘关系的远近和生长环境的差异,分子标记的数量、连锁距离、与目标基因之间的连锁关系等诸多因素的影响。总之,GS无法完全取代 传统育种,但可以整合到育种程序中应用到多种不同的场景。例如,GS得到的候选材料可以与回交 等其他的育种策略相结合;GS也可以辅助把地方或外源品系的多个优异等位基因快速转到重要的核心种质中,快速创制新的育种亲本材料。未来GS预测模型的高效应用需要一个集多组学数据、分析流程、方法、应用场景、育种策略等的管理系统,需要融合遗传学、基因组学、生物信息学、大数据分析和人工智能等多学科知识和技术的应用。通过结合多种育种策略和GS模型、种质系谱、表型组和环境数据,重点利用已知基因功能信息,将这些信息全面整合到上述知识库中,将有助于突破GS在应用上的局限,发展新型分子设计育种技术,全面提高育种效率。
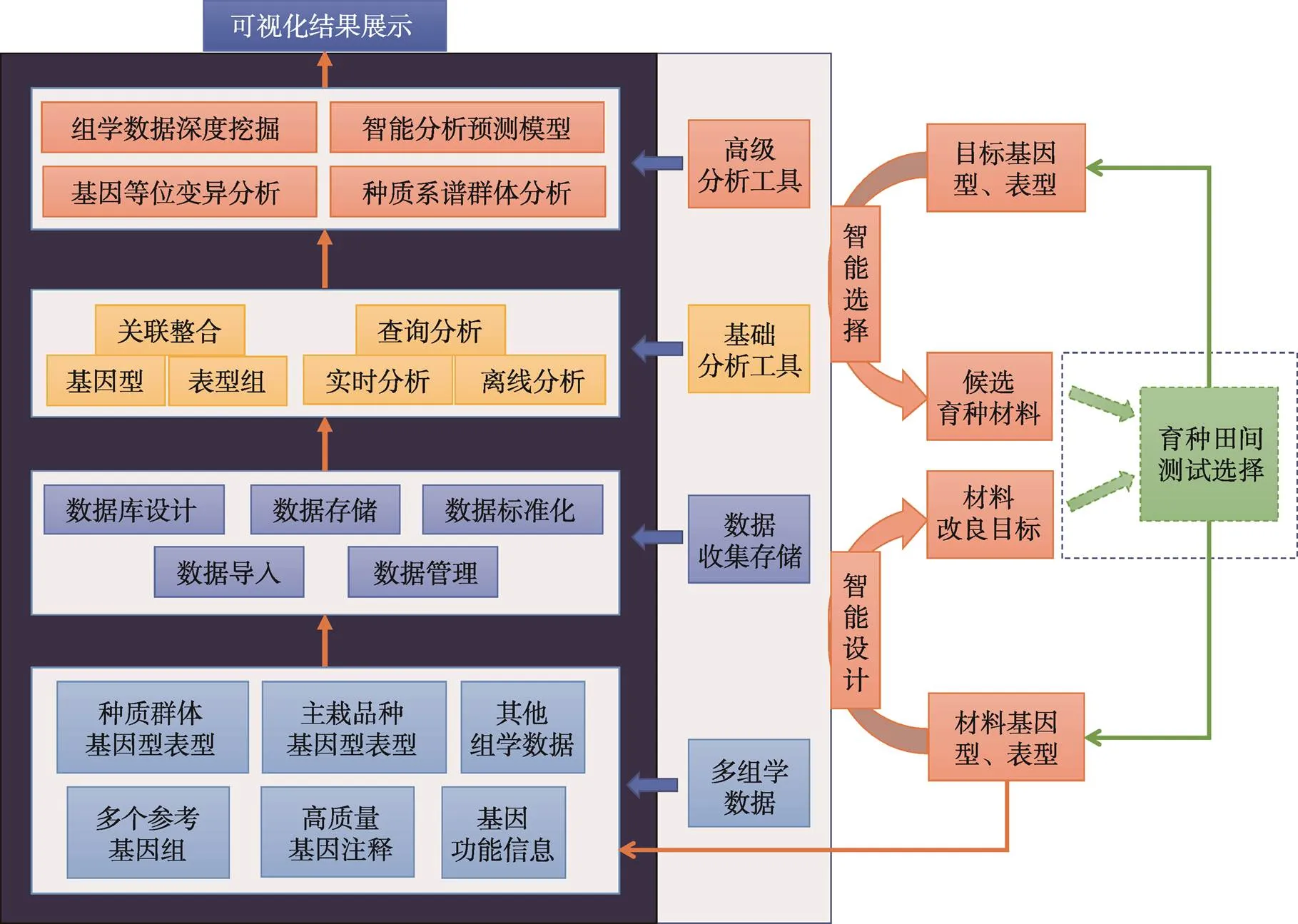
图2 作物整合组学知识库的数据、功能和应用示意图
知识库提供数据存储、管理、查询、可视化和智能分析等工具,育种材料将利用知识库的分析工具进行设计和筛选,同时材料的基因型和表型数据也持续上传到知识库中进行共享和重复利用。
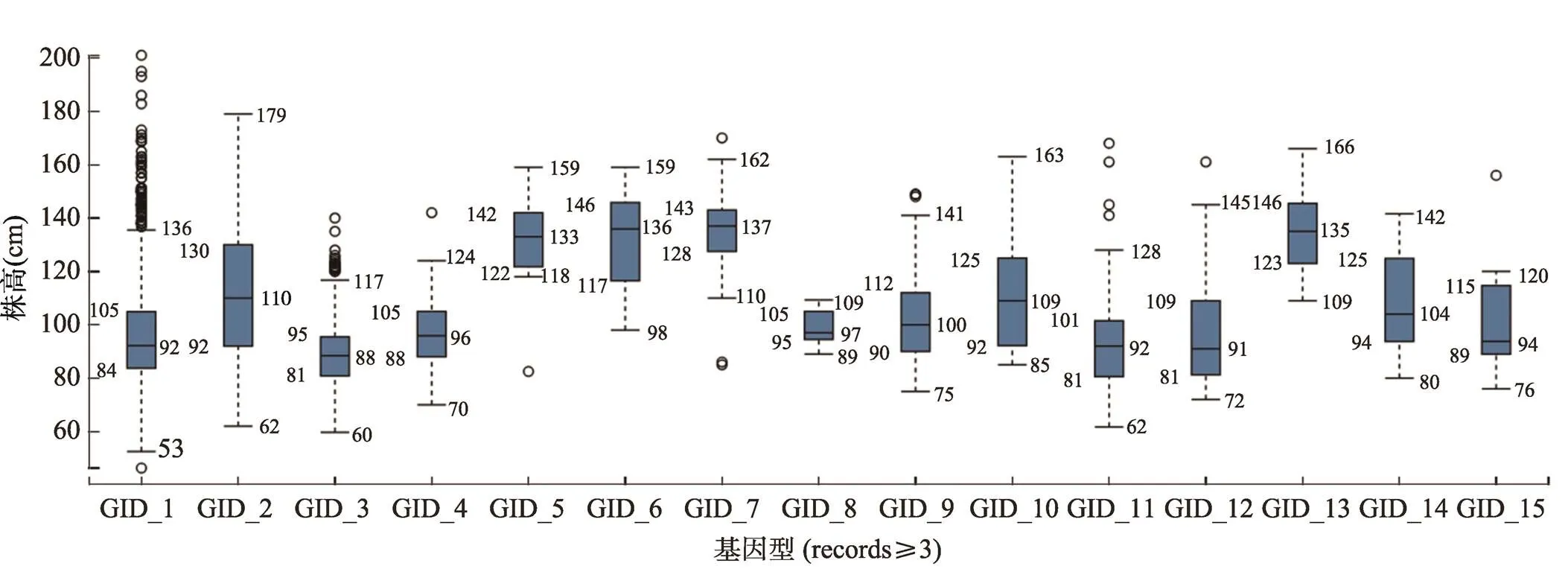
图3 群体中基因型跟表型值的关联示例
横坐标显示了目标基因在群体中出现频率最高的15个等位基因,纵坐标显示了每个等位基因在群体中对应的表型值(株高)统计。
近年来群体基因组研究发现,单个参考基因组无法代表一个物种的全部遗传成分,而是需要一个泛基因组(Pan-genome)来覆盖一个物种群体的全部基因及个体之间的有无变异序列[22]。例如,在两个水稻亚种日本晴和蜀恢498之间,有大量的有无结构变异[9];在水稻群体中,有大量的蛋白编码基因和其他序列在日本晴基因组上是缺失的[23]。在日本晴和蜀恢498之间有些直系同源(等位)基因DNA序列差异较大(但在蛋白质序列水平上则差异较小),导致用重测序的方法难以检测到这些同源基因的存在,而通过两个基因组序列的直接比较或基因注释的比较就能很容易发现这些遗漏的基因。此外,由于作物基因组的高复杂性,个体中的很多序列或结构变异往往都只存在于一些小的亚群体中。因此,具有亚群代表性的多个高质量的参考基因组对于研究一个物种的全部基因功能信息就变得非常必要。在育种应用上,因为GS等育种技术倾向于缩小遗传多样性,将地方种或近缘野生材料的优异等位基因聚合到育种群体中就成为一项非常重要的任务[24]。这些参考基因组或泛基因组以及亚群特异的基因功能信息能够促进对多样化遗传资源的分析,以及对这些材料中的优异等位基因的识别和利用。
目前,由于单分子长片段测序成本的持续降低,利用HERA结合其他开源软件,以很低的费用构建高质量作物基因组已经变得完全可行,甚至已变成了常规化工作,如在水稻中只需要不到3万元就能得到一个高质量(类似于日本晴)的基因组。在未来几年内,本课题组将致力于把大量的水稻、大豆、小麦等作物的高质量参考基因组和更多的群体基因型信息整合到已有的知识库中,同时进一步整合大群体的、系统性的、多环境下的表型组数据,以构建全面地从基因型到表型的全基因组预测模型。除了传统的人工测量的表型值,比如生育期、株高、分蘖数、穗粒数等,近年来发展起来的高通量田间表型组技术[25~30]利用传感器和光成像系统,结合实验室样品分析,以及高性能计算及图像分析技术和自动化分析软件的支撑,能够自动化获取多环境下包含几千个个体的关键生长时期、关键时间点、不同空间尺度上的变异的表型组学数据和环境数据。预期这些新型表型组数据在未来5~10年中将会呈现爆炸式增长。此外,该知识库还会整合高质量的基因注释、更多的基因组元件功能信息,并发展功能多样的用户友好的数据管理查询展示工具、多组学智能分析工具、基于基因型和表型进行育种材料选择和智能设计的软件工具等。相信随着这一多作物多组学整合知识库的建立和不断丰富完善,它必将为农作物功能基因组及分子设计育种的研究提供坚实的大数据和智能分析技术支撑。
[1] Zeng D, Tian Z, Rao Y, Dong G, Yang Y, Huang L, Leng Y, Xu J, Sun C, Zhang G, Hu J, Zhu L, Gao Z, Hu X, Guo L, Xiong G, Wang Y, Li J, Qian Q. Rational design of high-yield and superior-quality rice., 2017, 3: 17031.
[2] Meuwissen TH, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps., 2001, 157(4): 1819–1829.
[3] Wang JK, Li HH, Zhang XC, Yin CB, Li Y, Ma YZ, Li XH, Qiu LJ, Wan JM. Molecular design breeding in crops in China., 2011, 37(2): 191–201.王建康, 李慧慧, 张学才, 尹长斌, 黎裕, 马有志, 李新海, 邱丽娟, 万建民. 中国作物分子设计育种. 作物学报, 2011, 37(02): 191–201.
[4] 顾铭洪, 刘巧泉. 作物分子设计育种及其发展前景分析. 扬州大学学报 (农业与生命科学版), 2009, 30(1): 64–67.
[5] 苏岩, 钱前, 曾大力. 水稻分子设计育种的现状和展望. 中国稻米, 2010, 16(02): 5–9.
[6] Yu H, Wang B, Chen MJ, Liu GF, Li JY. Research advance and perspective of rice breeding by molecular design., 2018, 30(10): 1032–1037.余泓, 王冰, 陈明江, 刘贵富, 李家洋. 水稻分子设计育种发展与展望. 生命科学, 2018, 30(10): 1032–1037.
[7] 薛勇彪, 韩斌, 种康, 王台, 何祖华, 傅向东, 储成才, 程祝宽, 徐云远, 李明. 水稻分子模块设计研究成果与展望. 中国科学院院刊, 2018, 33(09): 900–908.
[8] Wu B, Hu W, Xing YZ. The history and prospect of rice genetic breeding in China., 2018, 40(10):841–857.吴比, 胡伟, 邢永忠. 中国水稻遗传育种历程与展望. 遗传, 2018, 40(10): 841–857.
[9] Du H, Yu Y, Ma Y, Gao Q, Cao Y, Chen Z, Ma B, Qi M, Li Y, Zhao X, Wang J, Liu K, Qin P, Yang X, Zhu L, Li S, Liang C. Sequencing and de novo assembly of a near complete indica rice genome., 2017, 8: 15324.
[10] Zhang L, Li X, Ma B, Gao Q, Du H, Han Y, Li Y, Cao Y, Qi M, Zhu Y, Lu H, Ma M, Liu L, Zhou J, Nan C, Qin Y, Wang J, Cui L, Liu H, Liang C, Qiao Z. The tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance., 2017, 10(9): 1224– 1237.
[11] Ling HQ, Ma B, Shi X, Liu H, Dong L, Sun H, Cao Y, Gao Q, Zheng S, Li Y, Yu Y, Du H, Qi M, Li Y, Lu H, Yu H, Cui Y, Wang N, Chen C, Wu H, Zhao Y, Zhang J, Li Y, Zhou W, Zhang B, Hu W, van Eijk MJT, Tang J, Witsenboer HMA, Zhao S, Li Z, Zhang A, Wang D, Liang C. Genome sequence of the progenitor of the wheat A subgenome., 2018, 557(7705): 424–428.
[12] Li M, Zhang D, Gao Q, Luo Y, Zhang H, Ma B, Chen C, Whibley A, Zhang Y, Cao Y, Li Q, Guo H, Li J, Song Y, Zhang Y, Copsey L, Li Y, Li X, Qi M, Wang J, Chen Y, Wang D, Zhao J, Liu G, Wu B, Yu L, Xu C, Li J, Zhao S, Zhang Y, Hu S, Liang C, Yin Y, Coen E, Xue Y. Genome structure and evolution ofL., 2019, 5(2): 174–183.
[13] Du, H, Liang C. Assembly of chromosome-scale contigs by efficiently resolving repetitive sequences with long reads., 2018, doi: 10.1101/345983.
[14] Liang C, Mao L, Ware D, Stein L. Evidence-based gene predictions in plant genomes., 2009, 19(10): 1912–1923.
[15] Yu, H, Lu, L, Jiao, B, Liang, C. Systematic discovery of novel and valuable plant gene modules by large-scale RNA-seq samples., 2019, 35(3): 361–364.
[16] Huang X, Han B. Natural variations and genome-wide association studies in crop plants., 2014, 65: 531–551.
[17] Zhou Z, Jiang Y, Wang Z, Gou Z, Lyu J, Li W, Yu Y, Shu L, Zhao Y, Ma Y, Fang C, Shen Y, Liu T, Li C, Li Q, Wu M, Wang M, Wu Y, Dong Y, Wan W, Wang X, Ding Z, Gao Y, Xiang H, Zhu B, Lee SH, Wang W, Tian Z. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean., 2015, 33(4): 408–414.
[18] Fang C, Ma Y, Wu S, Liu Z, Wang Z, Yang R, Hu G, Zhou Z, Yu H, Zhang M, Pan Y, Zhou G, Ren H, Du W, Yan H, Wang Y, Han D, Shen Y, Liu S, Liu T, Zhang J, Qin H, Yuan J, Yuan X, Kong F, Liu B, Li J, Zhang Z, Wang G, Zhu B, Tian Z. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean., 2017, 18(1): 161.
[19] Desta ZA, Ortiz R. Genomic selection: genome-wide prediction in plant improvement., 2014, 19(9): 592–601.
[20] Crossa J, Pérez-Rodríguez P, Cuevas J, Montesinos-López O, Jarquín D, de Los Campos G, Burgueño J, González- Camacho JM, Pérez-Elizalde S, Beyene Y, Dreisigacker S, Singh R, Zhang X, Gowda M, Roorkiwal M, Rutkoski J, Varshney RK. Genomic selection in plant breeding: methods, models, and perspectives., 2017, 22(11): 961–975.
[21] Messina CD, Technow F, Tang T, Totir R, Gho C, Cooper M. Leveraging biological insight and environmental variation to improve phenotypic prediction: Integrating crop growth models (CGM) with whole genome prediction (WGP)., 2018, 100: 151–162.
[22] Tao Y, Zhao X, Mace E, Henry R, Jordan D. Exploring and exploiting Pan-genomics for crop improvement,, 2019, 12(2): 156–169.
[23] Wang W, Mauleon R, Hu Z, Chebotarov D, Tai S, Wu Z, Li M, Zheng T, Fuentes RR, Zhang F, Mansueto L, Copetti D, Sanciangco M, Palis KC, Xu J, Sun C, Fu B, Zhang H, Gao Y, Zhao X, Shen F, Cui X, Yu H, Li Z, Chen M, Detras J, Zhou Y, Zhang X, Zhao Y, Kudrna D, Wang C, Li R, Jia B, Lu J, He X, Dong Z, Xu J, Li Y, Wang M, Shi J, Li J, Zhang D, Lee S, Hu W, Poliakov A, Dubchak I, Ulat VJ, Borja FN, Mendoza JR, Ali J, Li J, Gao Q, Niu Y, Yue Z, Naredo MEB, Talag J, Wang X, Li J, Fang X, Yin Y, Glaszmann JC, Zhang J, Li J, Hamilton RS, Wing RA, Ruan J, Zhang G, Wei C, Alexandrov N, McNally KL, Li Z, Leung H. Genomic variation in 3, 010 diverse accessions of Asian cultivated rice., 2018, 557(7703): 43–49.
[24] Zhang GQ. Research progress on breeding by design based on the SSSL library in rice., 41(8): 754–760.张桂权. 基于SSSL平台的水稻设计育种研究进展. 遗传, 41(8): 754–760.
[25] Araus JL, Cairns JE. Field high-throughput phenotyping: the new crop breeding frontier., 2014, 19(1): 52–61.
[26] Pan YH. Analysis of concepets and categories of plant phenome and phemomics., 2015, 41(02): 175–186.潘映红. 论植物表型组和植物表型组学的概念与范畴. 作物学报, 2015, 41(02): 175–186.
[27] Mu JH, Chen YZ, Feng H, Li WJ, Zhou LB. A new revolution in crop Breeding: the era of high-throughput phemomics., 2016, 34(06): 962–971.穆金虎, 陈玉泽, 冯慧, 李文建, 周利斌. 作物育种学领域新的革命: 高通量的表型组学时代. 植物科学学报, 2016, 34(06): 962–971.
[28] Duan LF, Yang WN. Research advances and future scenarios of rice phenomics., 2016, 28(10): 1129–1137.段凌凤, 杨万能. 水稻表型组学研究概况和展望. 生命科学, 2016, 28(10): 1129–1137.
[29] Zhou J, Tardieu F, Pridmore T, Doonan J, Reynolds D, Hall N, Griffiths S, Cheng T, Zhu Y, Wang XE, Jiang D, Ding YF. Plant phenomics: history,present status and challenges., 2018, 41(04): 580–588.周济, Francois Tardieu, Tony Pridmore, John Doonan, Daniel Reynolds, Neil Hall, Simon Griffiths, 程涛, 朱艳, 王秀娥, 姜东, 丁艳锋. 植物表型组学: 发展、现状与挑战. 南京农业大学学报, 2018, 41(04): 580–588.
[30] Rebetzke GJ, Jimenez-Berni J, Fischer RA, Deery DM, Smith DJ. Review: High-throughput phenotyping to enhance the use of crop genetic resources., 2019, 282: 40–48.
From genome analysis to construction of an integrated omics knowledgebase for crops
Chengzhi Liang1,2
The advances in high-throughput technologies have enabled high-speed accumulation of omics data, which contain a large amount of genetic variations and their functional information. The integration and deep utilization of those data will be a long-term and difficult task, which requires highly efficient data storage and powerful data analysis and mining tools. In the past several years, our group has conducted multi-level genomic analyses in several plants, including genome assembly and annotation, comparative and population genomic studies, through collaboration with other labs inside and outside of our institution. Meanwhile, we have integrated a large amount of rice germplasm information and omics data into a structural database and developed related data query, visual display and mining web tools. Here, we summarize some of those results and discuss our next goal to construct an integrated omics knowledgebase for crops to support functional genomics and molecular design breeding.
genome analysis; database; big omics data; molecular design breeding; crop; omics knowledgebase
2019-06-14;
2019-08-12
中国科学院重点部署项目(编号:ZDRW-ZS-2019-2-0105)资助[Supported by the Key Program of Chinese Academy of Sciences (No. ZDRW-ZS-2019-2-0105)]
梁承志,博士,研究员,研究方向:基因组大数据分析。E-mail: cliang@genetics.ac.cn
10.16288/j.yczz.19-121
2019/9/2 16:21:24
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190902.1620.002.html
(责任编委: 刘宝)
