微生物组数据分析方法与应用
2019-09-24刘永鑫秦媛郭晓璇白洋
刘永鑫,秦媛,3,郭晓璇,白洋,3
微生物组数据分析方法与应用
刘永鑫1,2,秦媛1,2,3,郭晓璇1,2,白洋1,2,3
1. 中国科学院遗传与发育生物学研究所,植物基因组学国家重点实验室,北京 100101 2. 中国科学院遗传与发育生物学研究所,中国科学院–英国约翰英纳斯中心植物和微生物科学联合研究中心,北京 100101 3. 中国科学院大学现代农学院,北京 100049
高通量测序技术的发展衍生出一系列微生物组(microbiome)研究技术,如扩增子、宏基因组、宏转录组等,快速推动了微生物组领域的发展。微生物组数据分析涉及的基础知识、软件和数据库较多,对于同领域研究者开展学习和选择合适的分析方法具有一定困难。本文系统概述了微生物组数据分析的基本思想和基础知识,详细总结比较了扩增子和宏基因组分析中的常用软件和数据库,并对高通量数据下游分析中常用的几种方法,包括统计和可视化、网络分析、进化分析、机器学习和关联分析等,从可用性、软件选择以及应用等几个方面进行了概述。本文拟通过对当前微生物组主流分析方法的整理和总结,为同领域研究者更方便、灵活的开展数据分析,快速选择研究分析工具,高效挖掘数据背后的生物学意义提供参考,进一步推动微生物组研究在生物学领域的发展。
微生物组;数据分析;扩增子;宏基因组;分析流程
微生物组(microbiome)是指包括细菌、古菌、低(高)等真核生物、病毒等微生物的基因和基因组,及其周围环境在内的全部[1]。研究表明微生物组在人类和动植物的营养吸收[2]、疾病抵抗[3]和环境适应中起重要作用[4,5]。
近年来第二代测序(next generation sequencing, NGS)技术的发展使得基于非培养方法研究微生物组成为可能,并推动了微生物组研究进入了黄金发展时期[6]。目前对微生物组样本的研究主要集中在3个层面(图1A):(1)微生物培养层面:培养组学(Culturome)是该层面最重要的研究手段。通过在固体培养皿挑单菌落或使用96孔板液体高通量培养的方式获得微生物群落中可培养的菌落,随后结合标记基因(marker gene)测序、分离纯化等方法进行菌种鉴定和保藏。目前该方法已在人类[7]、拟南芥()[8]、水稻()[9]等物种中应用和报道;(2) DNA层面:针对DNA易于提取和保存的特点,研究者相继发展出扩增子(amplicon)、宏基因组(metagenome)[10]和宏病毒组(metavirome)等测序研究手段[11]。扩增子测序常用的标记基因主要包括原核生物的16S rRNA基因、真核生物的18S rRNA基因以及转录间隔区(internal transcribed spacers, ITS)等。由于扩增子测序仅能获得研究对象的物种组成信息,要想进一步研究物种所携带的其他功能基因,就需要开展宏基因组测序和分析;(3) mRNA层面:通过对微生物组样本提取RNA进行宏转录组(metatranscriptome)测序,可以根据微生物组样本中的基因表达谱进一步揭示微生物群落原位功能[12]。病毒包括DNA和RNA病毒两大类,想要全面开展宏病毒组学研究需要宏基因组结合宏转录组测序(图1A)。
鉴于微生物组编码的基因近千万[13],想要从微生物组海量数据中挖掘有效信息,必须了解和掌握本领域相关软件和数据库的使用,才能在计算机或服务器上开展可重现(reproducible)的数据分析。而传统的生物学家由于生物信息学知识相对薄弱、微生物组数据分析经验不足等情况,在数据分析过程中经常会面临Linux使用、代码重用和软件选择等众多困难。本文系统概述了当前微生物组数据主流分析的基本思路和步骤,同时对开展微生物组数据分析提供了建议,最后对本领域常用分析方法的优缺点和适用范围进行总结,以期对同行更高效地开展微生物组数据分析,挖掘大数据背后的生物学规律有所帮助。
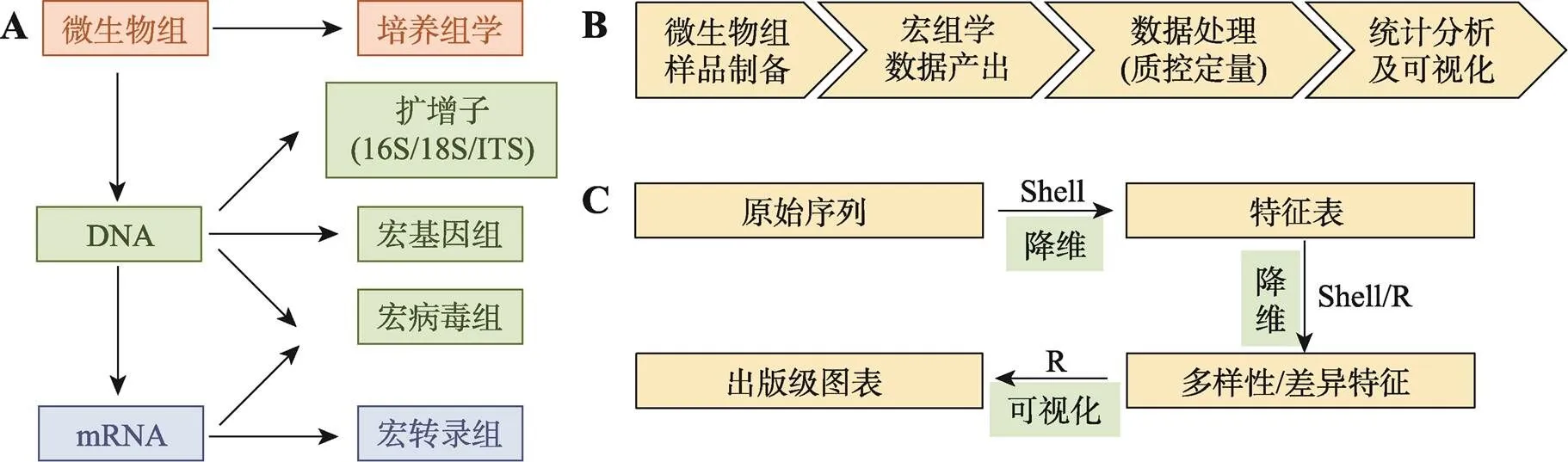
图1 微生物组研究方法概述
A:微生物组常用的研究层面和对应方法。微生物组按研究层面主要分为微生物培养、DNA和mRNA等3个层面;按研究技术主要包括培养组学(culturome)、扩增子(amplicon)、宏基因组(metagenome)、宏病毒组(metavirome) 和宏转录组(metatranscriptome)等测序技术[1,12]。B:微生物组研究的基本步骤。基于测序技术为基础的微生物组研究,主要分为样本制备、测序、数据处理和统计分析4个阶段。C:微生物组数据分析的基本步骤、常用环境和思想。组学数据分析主要分3步,图中箭头上描述了实现分析的常用语言环境Shell和/或R;图中箭头下展示各步分析的目的,即通过降维和可视化的基本思想,实现将大数据转化为可读图表。
1 微生物组数据分析的基本步骤
微生物组研究主要分为4个阶段(图1B):(1)微生物组样品制备:基于科学的实验设计,采集来自人、动植物或环境中的微生物组样本,并根据研究的目的,选择提取DNA或RNA等;(2)宏组学(meta- omics)数据产出:抽提样品的DNA或RNA后,通过构建测序文库和进行高通量测序来获得宏组学数据。例如,扩增子16S rRNA基因片段主要采用双端250 bp (pair-end 250 bp, PE250)测序,单个样本3~5万条序列的深度;宏基因组多采用PE150测序,获得微生物部分至少2千万条序列(150 bp´2´20 Mb = 6 Gb);(3)数据处理(质控定量):当获得微生物组数据后,首先要进行质量控制,包括去除测序和建库过程中人为添加的引物、接头以及测序过程中产生的低质量序列等。此外,宿主相关的微生物组测序结果中含有大量宿主序列,需采用比对宿主基因组的方式去除。获得的纯净序列(clean data)再比对至参考数据库或从头()组装的参考基因集,定量为特征表(feature table),根据序列注释类型可将特征表分为物种或功能基因组成表;(4)统计分析和可视化:特征表还需要进一步结合样本元数据(metadata)进行统计分析,并选择合适的图形进行可视化,有利于生物学规律的观察和总结,提高结果的可读性和传播性(图1B)。本文将主要对第3和第4步骤进一步讨论和总结。
当获得微生物组原始数据后,如何对其进行分析至高可读性的出版级别图表?为便于理解,本文将微生物组数据分析过程划分为3个主要步骤(图1C):
第一步:原始数据转换为特征表。微生物组数据通常为NGS产生的fastq格式序列文件,包括碱基序列和质量值,序列数量级可达106~109条。这就需要在高效的Shell环境下使用命令行工具对大数据进行质控和定量,降维至数量级为103~105的特征表。特征表常为计数型数据(count data),如物种分类学(taxonomy)表、可操作分类单元(operational taxonomic unit, OTU)表、扩增序列变异(amplicon sequence variant, ASV)表、基因丰度(gene abundance)表和通路丰度(pathway abundance)表等。
第二步:特征表转换为多样性和/或差异特征。例如,微生物组研究中扩增序列变异表和基因丰度表仍然很大,因此研究者常采用Alpha或Beta多样性分析、物种或功能层级注释、差异比较等方法,将数据表进一步降维至101~103。该数据结果更方便研究者运用专业知识挖掘规律和解释生物学问题。
第三步:数据可视化为出版级图表。近年来可视化语言和工具的发展提高了数据挖掘和结果解读的效率,如折线柱、柱状图、箱线图、散点图和热图等的广泛使用,更易于帮助研究者发现数据中的规律(图1C)。
从微生物组数据分析的全过程中可以看出,降维和可视化是大数据分析的核心指导思想,即把数据降维至可读的数量,通过可视化分析方便同领域研究者阅读和传播。实现这两个过程主要涉及两种语言环境,即首先通过Linux系统中的Shell语言配合工具软件实现大数据分析和降维,然后利用R语言(https://www.r-project.org)实现基于特征表的统计和可视化。因此熟悉Shell和R这两门语言的基础操作即可满足研究者微生物组数据分析的绝大多数需求。当然,微生物组分析中也常涉及Perl、Python、Java等语言的使用,它们更多作为软件和脚本在Shell环境下运行,用户可以根据自己的基础和习惯选择不同的语言环境进行分析和可视化。
2 微生物组数据分析常用的环境
微生物组数据分析需要在专门的语言环境下开展,熟悉常用的语言环境能够帮助我们更好地利用现有工具开展数据分析。目前本领域的分析工具主要集中在Shell和R两种语言环境下运行。几乎所有的服务器都是Linux系统,默认的Shell环境自带上百个命令和Bioconda近万个生物信息软件可快速搭建各种分析流程[14]。R语言开源免费,官网CRAN (https://cran.r-project.org/)发布了14 767个统计和可视化包,Bioconductor (http://www.bioconductor.org)上更有1741个生物学专用包(数量统计截止2019年8月20日),可实现最灵活的统计分析。掌握这两门语言基础,可以高效地利用现有软件开展数据分析、统计和可视化。本文重点介绍Shell和R语言,是因为这两类语言环境下有非常多可利用的生物学软件(包),用户可以通过极少的代码串联现有工具来实现数据分析。特别是对于初级使用者来说,学习和应用相对更加便捷。
Shell语言是与Linux系统交互命令的合集,几乎所有的微生物组分析工具都有可以在Linux服务器的的Shell环境下运行,而在其他环境中搭建分析流程非常困难。如果用户的电脑为Windows系列,需要安装远程访问Linux服务器的软件,如XShell、putty或ssh secure shell等,这里推荐使用商业化开发且对学校免费的XShell。而Mac系统是类UNIX系统内核,系统自带的Terminal程序即可实现远程访问Linux。R语言自带图形界面RGui,可以实现交互式统计分析和可视化,RScript命令可在命令行下执行R脚本。近两年快速发展的集成开发环境RStudio (https://www.rstudio.com/),自2018年升级至1.1版后同时支持Shell和R脚本的编辑和运行。RStudio是跨平台软件,在Windows/ Mac/Linux上都可以轻松安装,还有服务器版本可以在网页中运行,保证不同终端无需安装任何额外程序,即可保持数据分析工作环境的一致性。对于初学数据分析的研究者来说,可通过学习RStudio来掌握数据分析、代码管理、程序调试、结果图片调整和保存等操作。
有了好用的分析代码管理工具,还需要学习语言基础读懂分析代码,才能使用和修改现有的分析流程和方法。对于以数据分析为主的研究者,建议系统学习Shell和R语言基础。Shell语言推荐学习《鸟哥的LINUX私房菜基础学习篇(第四版)》,其中Linux的基本命令、文件系统和Shell脚本编写可重点学习,服务器管理员还需要学习系统和用户管理等内容。R语言推荐学习《ggplot2:数据分析与图形艺术(第2版)》[15],该工具书对系统认识各种图形、了解绘图原理和实现数据可视化非常有帮助。此外,通过学习网络上相关研究者整理总结的的基础知识和代码注释,对于初学者以及偶尔使用数据分析的研究者来说,可能更具有针对性和时效性。
3 微生物组领域常用软件
近10年,随着高通量测序技术的发展和应用,微生物组研究领域的相关分析方法和工具也取得了快速发展,大量优秀的软件、流程和可视化工具相继发布,进一步推动了本领域的发展。
3.1 扩增子分析软件
扩增子分析是微生物组领域应用最广泛的技术,可以快速获悉研究对象中的微生物多样性。本文将重点介绍3款(mothur, QIIME和USEARCH)在近10年内发表且引用过万次的扩增子分析软件(图2),其他更多相关软件介绍详见表1。
(1) mothur:由美国密歇根大学的Patrick D. Schloss教授团队在2009年发布的首个扩增子分析流程[16]。它整合了之前发表的OTU定义软件DOTUR[17]、OTU差异比较工具SONS[18]以及其他可用工具,实现了第一套较完整的分析流程,让广大研究者开展扩增子分析成为可能(图2)。
(2) QIIME:2010年,美国科罗拉多大学的Rob Knight教授(现单位美国加州大学圣地亚哥分校)团队发布QIIME (发音同chime)分析流程[19]。该流程可在Linux或Mac系统中运行,相比mothur具有更多的优点,主要包括:整合了200多款相关软件和包,实现每个步骤更多软件和方法的选择;提供150多个脚本,实现各种个性化分析,并可以应对不同类型数据和实验设计;流程开放程度高,容易整合新软件和方法;增强统计和可视化,实现多样性、物种组成、差异比较和网络等众多方法和出版级图表绘制。由于QIIME允许同领域研究者较自主地开展扩增子数据的个性化分析和可视化,逐渐成为本领域最受欢迎的软件(图2)。为了满足日益增长的测序数据量和可重复计算的要求,Gregory J. Caporaso教授于2016年起发起了基于Python 3语言从头编写的QIIME 2项目[20]。该项目实现了分析流程的可追溯以满足科研可重复计算的要求;同时推出了一系列新算法,如基于进化距离的快速算法条型(Striped) UniFrac[21]、物种分类新方法2-feature-classifier[22]等;更重要的是软件的可扩展性和得到了同际同行的广泛支持,如接头和引物序列去除工具cutadapt[23]、序列质量控制R包DADA2[24]、聚类和去冗余的软件VSEARCH[25]、纵向和成对样本分析工具longitudinal[26]等,甚至包括宏基因组、宏代谢组分析和中文帮助文档,极大了提高了流程的适用范围和易用性。
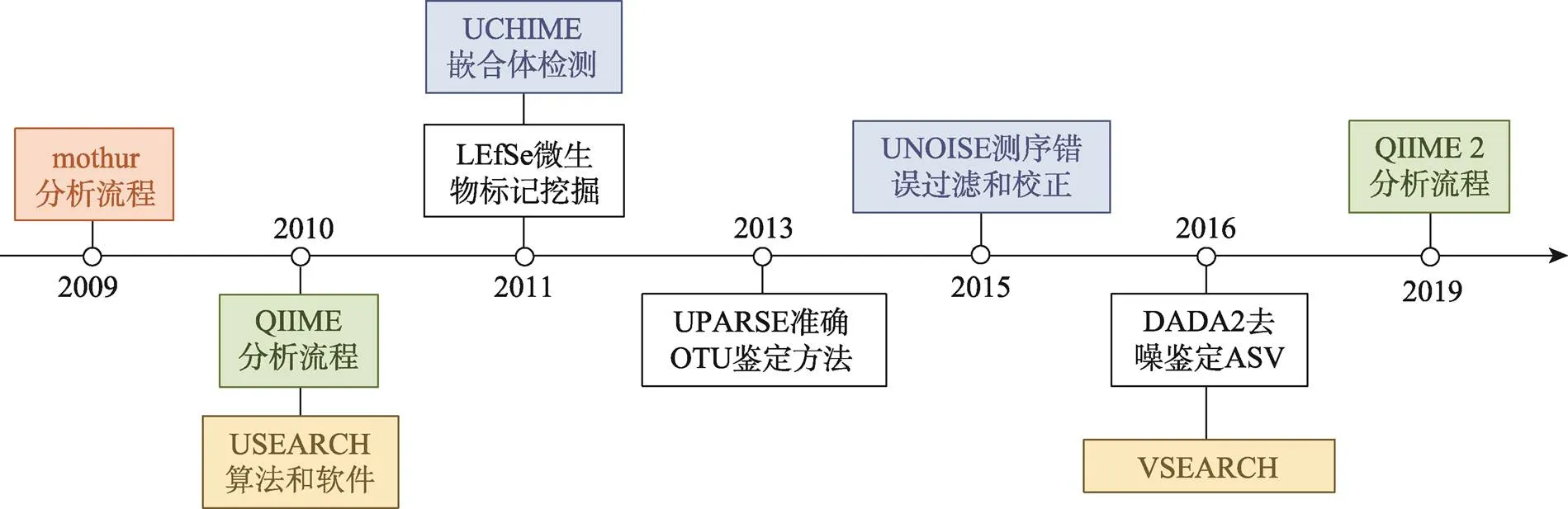
图2 近10年来微生物组领域的重要软件和算法
图中橙色为Patrick D. Schloss教授开发的分析流程mothur,绿色为Rob Knight教授主持开发的QIIME系列分析流程,蓝色显示Robert Edgar独立研究员编写的相关软件和算法。
续表
(3) USEARCH-based的扩增子分析流程。虽然已经发布了两套较完整的扩增子分析流程,但研究中存在的诸多问题却仍没有很好的解决。物理学背景的生物信息学家、独立研究员Robert Edgar在本领域开发了一系列经典的算法和软件,如高速序列比对软件USEARCH[27]、嵌合体检测软件UCHIME[28]、OTU代表性序列鉴定算法UPARSE[29]和测序数据错误过滤和去噪算法UNOISE等[30]。这些算法和软件的推出,极大的提高了扩增子数据分析的速度和准确度。在以上算法和软件的基础上,Robert逐渐将USEARCH发展成为包括近200种命令的完整扩增子分析流程,而且跨平台、体系小巧、无依赖关系和容易安装,其32位版本免费,64位版商业版和非赢利版分别售价1485和885美元,条件允许的实验室推荐购买,软件分析速度快且易用性强,可有效降低入门学习成本并节约宝贵时间。同时也有USEARCH类似的工具推出,如64位完全免费的VSEARCH[25],可实现USEARCH的核心功能,但下游分析功能略少。
从使用难易程度看,推荐初涉扩增子分析人员从使用USEARCH[25]或VSEARCH[25]开始,这两款软件允许用户在Windows或Mac笔记本上完成多达几百个样本分析项目。对于有一定基础且有Linux服务器的研究者,可进一步学习QIIME 2来实现更多种类的分析方法。
统计分析和可视化部分的工作常在R语言中实现。扩增子数据分析常用R包有vegan[31]、phyloseq[32]和microbiome[33]。vegan是群落生态包,可实现多样性、主坐标等分析,在微生物生态领域有广泛应用,甚至发展出了基于ggplot2版本的ggvegan[31]。phyloseq[32]包的功能主要包括多样性分析、差异比较和可视化等。针对没有R使用经验的用户,phyloseq还推出了网页版工具shiny-phyloseq[34],在浏览器中即可实现扩增子数据交互式分析。microbiome 包[33]包括多样性、核心OTU、物种组成、相关性和格式转换等80余个分析函数,提高微生物组分析的工作效率。
3.2 宏基因组分析软件
近年来,鸟枪法宏基因组(shotgun metagenomic)测序随着通量提高和价格下降得到了进一步发展,随之而来的是大量相关软件的研发和发表(表2)。较扩增子测序相比,宏基因组测序不仅能获得无偏的物种组成,还得获得研究对象的功能组成,甚至能拼接出部分微生物的基因组草图。
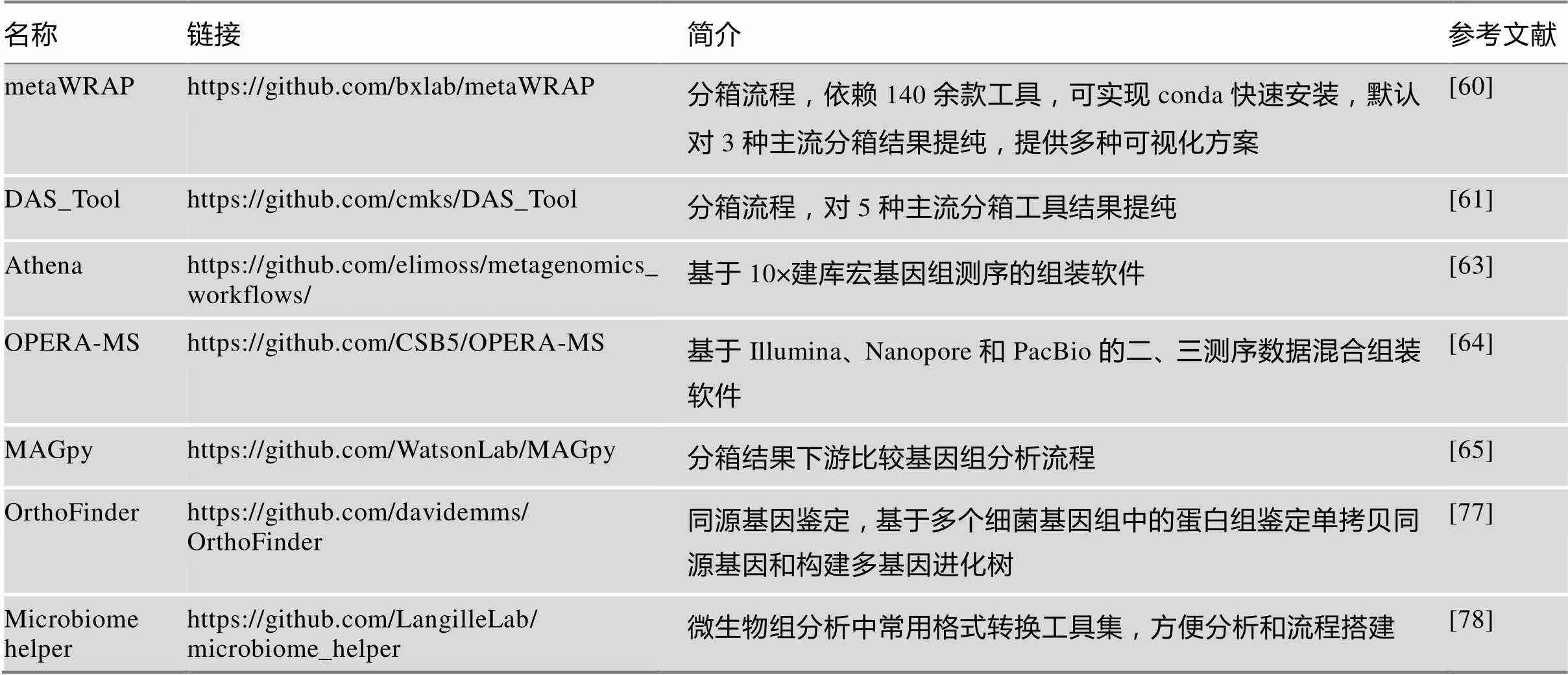
对于人类肠道这类研究较多的领域,可选择基于参考数据库比对快速实现宏基因组物种和功能组成定量的分析方案,如MetaPhlAn2[47]、Kraken2[48]实现序列的物种分类,HUMAnN2[49]实现功能组成定量。对于缺少高质量宏基因组参考数据库的领域,则需要从头()拼接宏基因组数据,并进行基因预测。常用的宏基因组拼接软件有MEGAHIT[70]和metaSPAdes[50]等,基因注释软件如Prokka[51]和GeneMarkS-2[52]等(表2)。对于多样品或多批次的宏基因组数据进行合并分析,通常还要采用CD-HIT[53]构建非冗余基因集(non-redundancy gene catalog),实现将所有样本基于统一的参考序列进行定量和比较。获得的基因集比对至多种蛋白功能注释数据库,提供更多角度观察数据的生物学意义,如常用的数据库有碳水化合物基因数据库CAZy[54]、抗生素抗性基因综合数据库CARD[55]和毒力因子数据库VFDB[56]等。
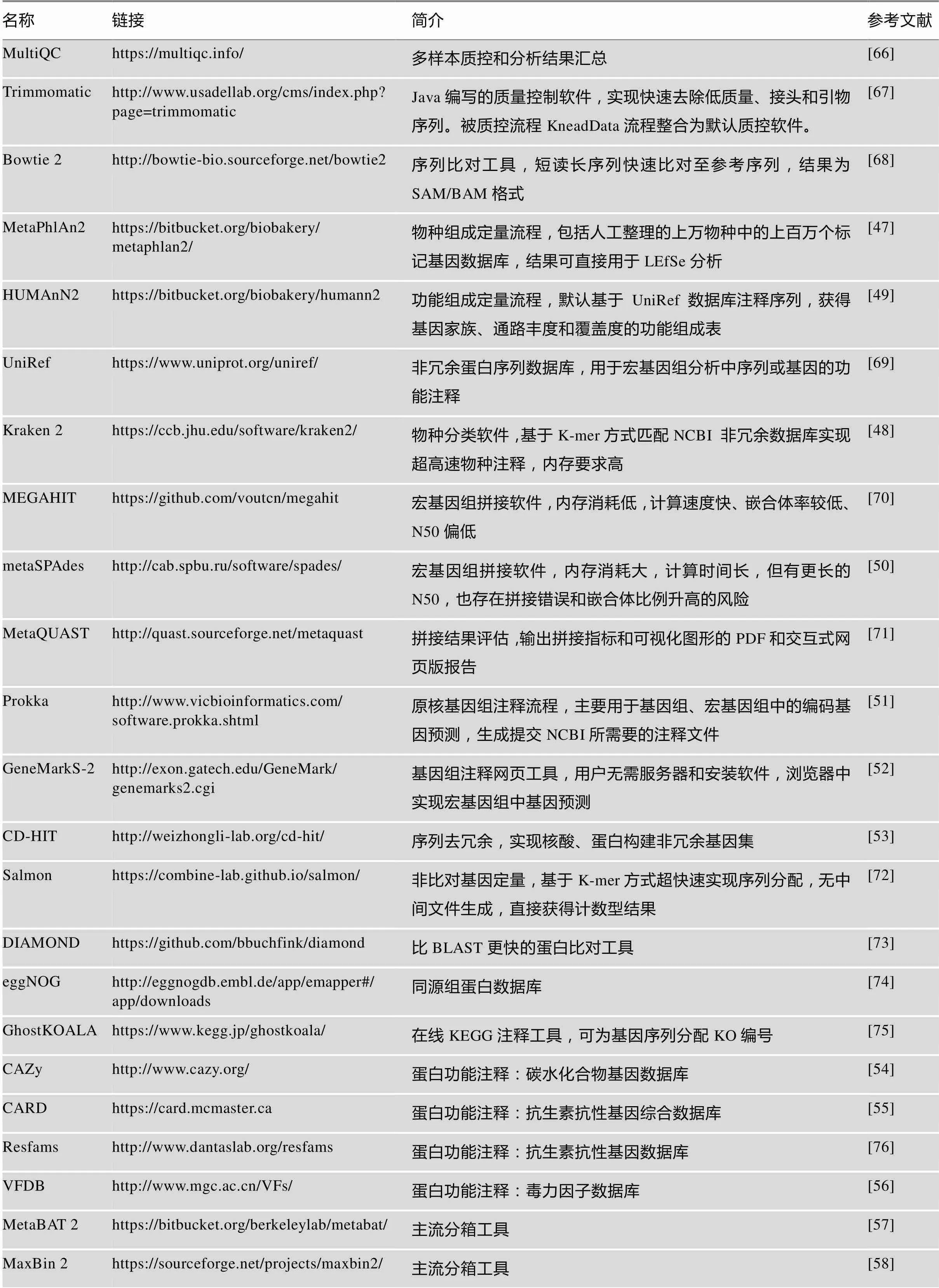
表2 宏基因组分析常用软件和数据库
续表
宏基因组测序除了可以揭研究对象的物种和功能组成外,还可能通过分箱(binning)方法组装出单菌基因组。近年来分箱软件快速发展,使获得不可培养微生物的基因组成为可能。目前常用的分箱工具有MetaBAT 2[57]、MaxBin 2[58]和CONCOCT[59]等,但结果差别较大。去年发表了两款分箱提纯工具metaWRAP[60]和DAS_Tool[61]解决了分箱工具选择难、结果差异大的问题,他们通常整合3~5款分箱工具的结果,进一步筛选和综合利用,获得更高质量的单菌基因组,同时提供分箱的定量、注释等一系列常用分析功能。值得注意的是,分箱获得的单菌基因组存在着不完整和高污染等问题,因此想要提高宏基因组中单菌组装的完整性,从实验手段进行改进并采用配套专用分析方法是未来的发展方向,如采用流式细胞术单细胞分选[62]、10×建库[63]、二三代混合测序[64,65]等新方法在宏基因组拼接和分箱中取得了较好的效果。宏基因组分析中常用的软件和数据库简介详见表2。
3.3 统计和可视化工具
扩增子和宏基因组分析获得的物种和功能组成表统称为特征表,是第二代测序数据分析结果中的通用格式,在下游分析中可以通过选择多种R包、图形化界面、命令行或网页版工具进行数据的转换和呈现。Bioconductor网站提供了上千种生物学数据分析R包,例如计数型数据可选基于负二项分布模型的差异统计R包edgeR[79]或DESeq2[80],组成型数据差异分析可选limma包[81],结合已知影响因素数据校正的差异比较可选支持广义线性混合效应模型的lme4包[82]。STAMP是为微生物组数据开发的跨平台、图形界面统计分析工具[83],可以实现主成分分析、多种统计方法进行两组或多组差异比较,结果可选散点图、箱线图、柱状图、热图和扩展柱状图等展示方法。LEfSe可以实现基于线性判别分析寻找特征向量的命令行工具[84],结果可选柱状图和基于GraPhlAn绘制的进化分枝图(cladogram)等展示方式[85],没有Linux服务器或不熟悉命令行工作的研究者还可以选择网页版LEfSe开展分析。此外,还有一些专门收集整理微生物组工具并提供在线分析和可视化的平台,让用户在浏览器中即可完成分析工作,例如MicrobiomeAnalyst[86]可实现基于特征表和元数据进行数据筛选、标准化、多样性分析、差异比较和机器学习等多种分析和可视化方案。
3.4 网络分析
网络分析是一门基于图论的学科,因其独特的视角和直观的可视式结果在微生物组数据分析中也有广泛的应用。2018年,发表综述文章系统介绍了目前主流网络分析方法的优缺点、适用范围和选择依据[87];发表综述文章介绍了网络图在群落结构研究中的作用和意义[88];此外,陈亮2017年在宏基因组公众号发布的《Co-occurrence网络图在R中的实现》对相关基础概念和具体的实现方法进行了介绍,也可供学习参考。常用的分析方法有网页工具MENAP[89],本地相似分析LSA[90]、专为微生物组稀疏型数据开发的相关性算法SPARCC[91]、作为Cytoscape[92]插件使用的CoNet[93]、R语言中的WGCNA[94]和SpiecEasi[95]包等。具体的操作也比较容易实现,例如在R语言环境中使用WGCNA[94]中包计算网络相关性质,采用igraph[96]包实现网络的可视化。对于网络的进一步分析、可视化细节调整,可将网络数据导入Cytoscape[92]或Gephi[97]中调整细节。目前该分析已在pH与微生物群落组装[98]、妊娠糖尿病与健康孕妇微生物组结构、洗牙后口腔微生物群落结构恢复等研究中得到应用[99,100]。
3.5 进化分析
微生物组数据非常适合开展进化分析,因为单物种的研究需要搜集和整理大量相关研究中的同源基因,而微生物组研究中的扩增子测序可获得的序列就是成千上万的同源基因,方便开展物种系统发育关系研究。进化分析主要分为多序列对齐、进化树构建和进化树美化等3个基本过程。由于微生物组中序列种类多且复杂度高,需要选择计算速度快的工具。多序列对齐可采用MAFFT[101]或MUSCLE[102];进化树构建可选FastTree[103]或IQ-TREE[104,105];最后采用Evolview[106]或iTOL[107]在线进行进化树的可视化和美化。推荐将序列对应的物种和丰度信息表使用R脚本table2itol (https://github.com/mgoeker/ table2itol)格式化为iTOL的输入文件。此外,R语言中的ggtree包也可以实现进化树的注释和美化[108]。展示物种注释层级结构的进化分枝图,推荐使用GraPhlAn进行可视化[85]。宏基因组测序是鸟枪法随机片段测序,进化分析需要采用OrthoFinder[77]基于分箱结果鉴定单拷贝同源基因,并构建多基因进化树。
3.6 机器学习
机器学习是当前计算机算法研究中最热门的领域,专门研究计算机如何模拟或实现人类的学习行为,以获取新的知识或技能,重新组织已有的知识结构使之不断改善自身的性能[109]。目前在微生物组领域常用的机器学习方法有随机森林(random forest)、支持向量机(support vector machine, SVM)和Adaboost等。其中随机森林分类(classification)在饮食习惯分型[110]、疾病诊断[111]、植物亚种预测[9]等领域有较多应用;随机森林回归(regression)在婴儿营养健康[2]、法医学[112]、时间序列预测[113]等领域有广泛的应用。开展随机森林分析可在R语言中通过使用randomForest包实现[114]。深度学习是机器学习领域新发展的方法,最近预印本服务器BioRxiv在线发表了基于肠道菌群数据的深度学习可准确预测人类真实年龄[115],此项研究还被杂志新闻报导。
3.7 其他分析工具
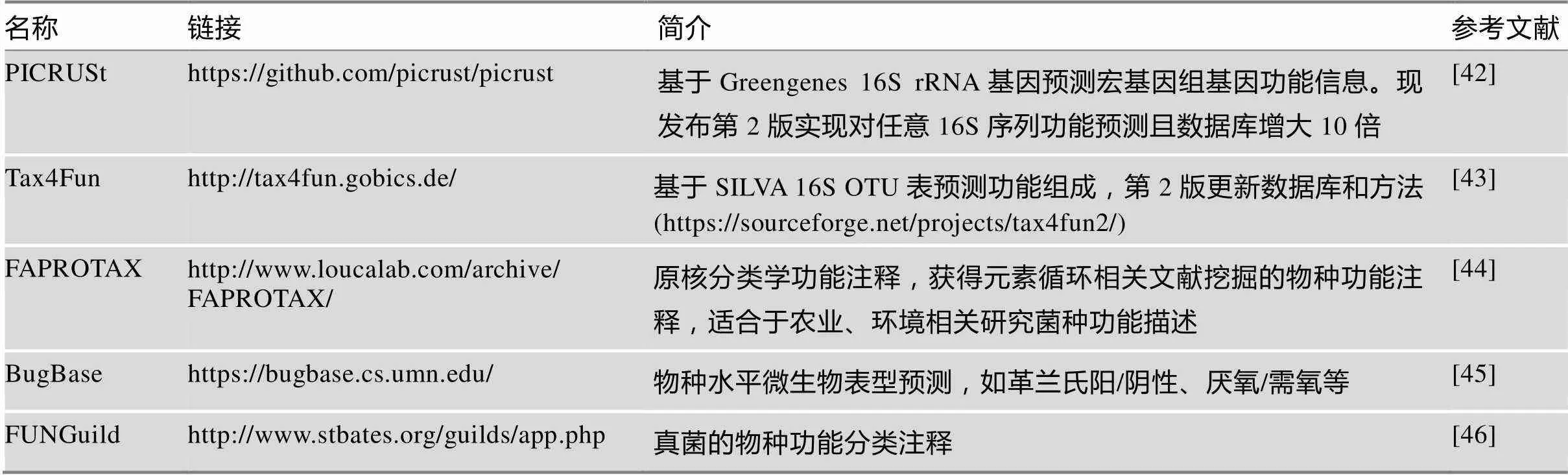
许多其他领域的分析方法在微生物组中也得到了推广和应用。全基因组关联分析(genome-wide association study, GWAS)[116]在鉴定人类疾病相关基因中发挥了巨大作用,目前也应用于微生物组领域来大规模探索人类与微生物组间的调控规律[117,118]、植物微生物组与产量[119]等。环境因子关联分析也有较多的分析方法在微生物生态学中得到广泛应用,如揭示温度[120]、pH[121]和盐分[122]等在不同环境中是微生物群落结构的决定因素。更多关于微生物组下游分析工具的介绍,详见表3。
4 分析代码重用
很多文章中的分析和可视化结果并非基于发表软件,而且作者自编程实现的分析。如果想参考文章中的分析方法和图表,根据方法描述自行组合工具或编写代码是非常有挑战的工作。目前很多文章发表时提供了分析代码,链接位于文章“代码可用(Code Available)”栏目,代码保存于Github等代码备份网站。基于文章作者分享的代码和测试数据,更容易重复文章中发表的分析方法,在理解的基础上替换为自己的数据开展分析,甚至可在源代码基础上修改分析方案,获得更合理的结果。分析代码的重现性在研究中可极大地提高工作效率,节省研究者大量开发分析代码的时间。表4列举了一些提供可重复分析代码的实验室,供研究者参考。
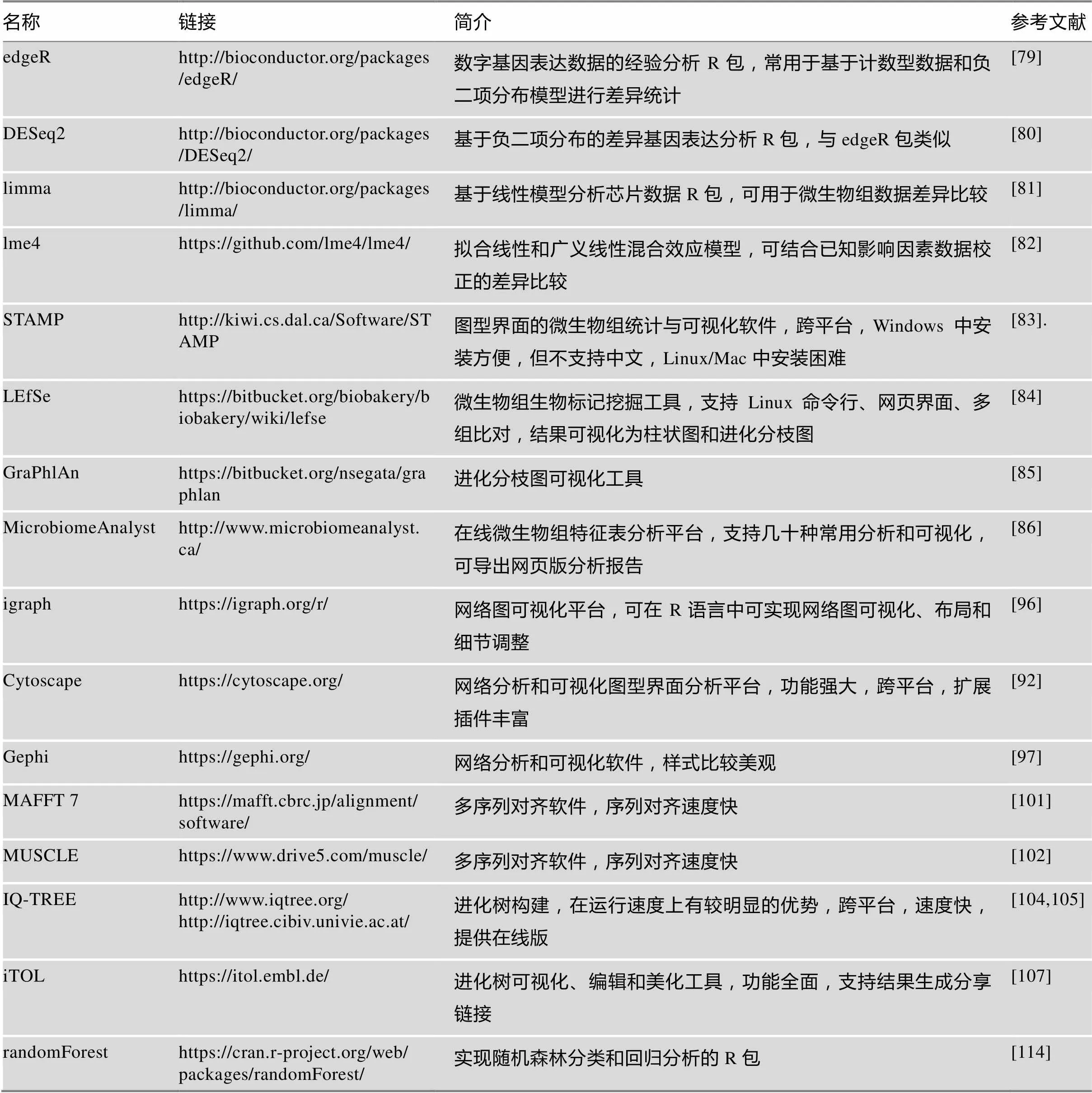
表3 微生物组下游通用分析工具
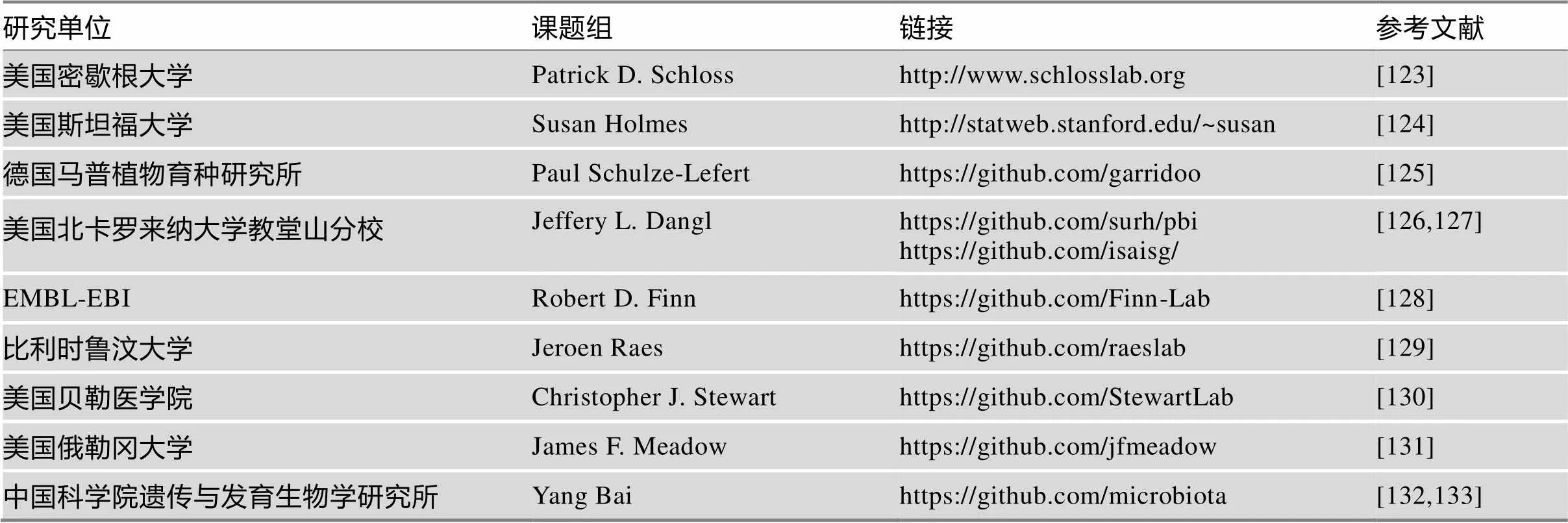
表4 部分提供统计分析代码的实验室
5 结语与展望
近10年来,第二代测序技术通量的提高和价格的下降,极大地推动了微生物组领域的发展,使得研究者拓宽了微生物组研究对象的深度和广度,揭示了极端环境、植物、动物、人类肠道、海洋、土壤等领域的微生物组成和功能[6]。目前宏基因组研究主要以短读长的Illumina Seq/Nova系列或华大基因的BGI Seq系列平台产出数据为主,虽然获得数据通量大,但数据拼接质量仍有较大提升空间。近年来,Pacific BioSciences (PacBio)和Oxford Nanopore Technologies (ONT)等三代测序技术快速发展,虽然受到测序错误率高和配套软件缺乏的困扰,但在读长、测序速度等方面的优势正在逐渐突显。Charalampou等[134]应用ONT技术对患者呼吸道细菌宏基因组进行测序,实现了6 h内快速诊断致病菌。
目前微生物组研究中应用最广泛的是扩增子测序技术,该技术可以快速地揭示群落的微生物组成,而且具有操作简单、成本低、有效避免宿主污染、方便开展大规模研究等优势。但扩增子的研究范围仅限引物可扩增部分DNA的物种组成,而且受扩增基因拷贝数和多态性的影响,如果想进一步了解微生物组的全貌和功能基因,宏基因组是更有效的研究方法。宏基因组不仅可以无偏的获得研究对象中细菌、真菌、古菌、病毒和原生动物等一切以DNA为遗传物种的物种序列信息、确定其物种和功能组成,更有潜力获得未培养物种的功能基因,甚至是基因组草图。目前虽然已经有一些宏基因组分箱、分箱提纯的工具,但仍处于发展的初级阶段,还有很多有待改进的方向,如计算不同长度K-mer频率、比对参考数据库去除已知物种降低复杂度和/或结合三代长读长的测序数据等[64,135]。
提高微生物组数据分析的效率,高质量的参考数据库是基础,而这一领域的发展依赖于大规模培养组学的应用和更多高质量参考基因组的公布。同时,对发表数据的分类整理、提高可用性以及进一步挖掘也十分必要。例如,R包curatedMetagenomicData整理了46个研究中的8184个宏基因组样本,对超100 TB的原始数据采取了严格质控进而获得了相关物种和功能组成表,方便同领域研究者对数据进一步挖掘和查询[136];ML Repo数据库整理来自15篇文章中的33个人类微生物组IBD、糖尿病、肥胖和癌症等分类和年龄回归数据集,研究者可按类浏览下载这些数据,用于进一步挖掘和方法评估[137];意大利特伦托大学Nicola Segata团队利用来自不同地理位置、生活方式和年龄人群的9428个宏基因组,突破性地重建了15万个人体微生物基因组草图[138]。以上对发表数据整理和再利用的例子,为今后开发更多基于发表数据的数据库和分析工具提供了借鉴和参考。
[1] Marchesi JR, Ravel J. The vocabulary of microbiome research: a proposal.,2015, 3(1): 31.
[2] Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, Barratt MJ, VanArendonk LG, Zhang Q, Province MA, Petri WA Jr, Ahmed T, Gordon JI. Persistent gut microbiota immaturity in malnourished Bangladeshi children.,2014, 510: 417–421.
[3] Bai Y, Qian JM, Zhou JM, Qian W. Crop Microbiome: breakthrough technology for agriculture.,2017, 32(3): 260–265.白洋, 钱景美, 周俭民, 钱韦. 农作物微生物组:跨越转化临界点的现代生物技术. 中国科学院院刊,2017, 32(3): 260–265.
[4] Wang J, Jia H. Metagenome-wide association studies: fine-mining the microbiome.,2016, 14: 508–522.
[5] Xie JP, Han YB, Liu G, Bai LQ. Research advances on microbial genetics in China in 2015.,2016, 38(9): 765–790.谢建平, 韩玉波, 刘钢, 白林泉. 2015年中国微生物遗传学研究领域若干重要进展. 遗传,2016, 38(9): 765–790.
[6] White RA Ⅲ, Callister SJ, Moore RJ, Baker ES, Jansson JK. The past, present and future of microbiome analyses.,2016, 11: 2049–2053.
[7] Zou Y, Xue W, Luo G, Deng Z, Qin P, Guo R, Sun H, Xia Y, Liang S, Dai Y, Wan D, Jiang R, Su L, Feng Q, Jie Z, Guo T, Xia Z, Liu C, Yu J, Lin Y, Tang S, Huo G, Xu X, Hou Y, Liu X, Wang J, Yang H, Kristiansen K, Li J, Jia H, Xiao L. 1,520 reference genomes from cultivatedhuman gut bacteria enable functional microbiome analyses.,2019, 37(2): 179–185.
[8] Bai Y, Müller DB, Srinivas G, Garrido-Oter R, Potthoff E, Rott M, Dombrowski N, Münch PC, Spaepen S, Remus-Emsermann M, Hüttel B, McHardy AC, Vorholt JA, Schulze-Lefert P. Functional overlap of theleaf and root microbiota.,2015, 528(7582): 364–369.
[9] Zhang J, Liu YX, Zhang N, Hu B, Jin T, Xu H, Qin Y, Yan P, Zhang X, Guo X, Hui J, Cao S, Wang X, Wang C, Wang H, Qu B, Fan G, Yuan L, Garrido-Oter R, Chu C, Bai Y.is associated with root microbiota composition and nitrogen use in field-grown rice.,2019, 37(6): 676–684.
[10] Shi W, Li M, Wei G, Tian R, Li C, Wang B, Lin R, Shi C, Chi X, Zhou B, Gao Z. The occurrence of potato common scab correlates with the community composition and function of the geocaulosphere soil microbiome.,2019, 7(1): 14.
[11] Ma Y, You X, Mai G, Tokuyasu T, Liu C. A human gut phage catalog correlates the gut phageome with type 2 diabetes.,2018, 6(1): 24.
[12] Yu K, Yi S, Li B, Guo F, Peng X, Wang Z, Wu Y, Alvarez-Cohen L, Zhang T. An integrated meta-omics approach reveals substrates involved in synergistic interactions in a bisphenol A (BPA)-degrading microbial community.,2019, 7(1): 16.
[13] Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, Juncker AS, Manichanh C, Chen B, Zhang W, Levenez F, Wang J, Xu X, Xiao L, Liang S, Zhang D, Zhang Z, Chen W, Zhao H, Al-Aama JY, Edris S, Yang H, Wang J, Hansen T, Nielsen HB, Brunak S, Kristiansen K, Guarner F, Pedersen O, Doré J, Ehrlich SD, MetaHIT Consortium, Bork P, Wang J, Pons N, Le Chatelier E, Batto JM, Kennedy S, Haimet F, Winogradski Y, Pelletier E, LePaslier D, Artiguenave F, Bruls T, Weissenbach J, Turner K, Parkhill J, Antolin M, Casellas F, Borruel N, Varela E, Torrejon A, Denariaz G, Derrien M, van Hylckama Vlieg JET, Viega P, Oozeer R, Knoll J, Rescigno M, Brechot C, M'Rini C, Mérieux A, Yamada T, Tims S, Zoetendal EG, Kleerebezem M, de Vos WM, Cultrone A, Leclerc M, Juste C, Guedon E, Delorme C, Layec S, Khaci G, van de Guchte M, Vandemeulebrouck G, Jamet A, Dervyn R, Sanchez N, Blottière H, Maguin E, Renault P, Tap J, Mende DR. An integrated catalog of reference genes in the human gut microbiome.,2014, 32(8): 834–841.
[14] Grüning B, Dale R, Sjödin A, Chapman BA, Rowe J, Tomkins-Tinch CH, Valieris R, Köster J, Bioconda Team. Bioconda: sustainable and comprehensive software distribution for the life sciences.,2018, 15(7): 475–476.
[15] Wickham H. ggplot2: elegant graphics for data analysis., 2016.
[16] Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities.,2009, 75(23): 7537–7541.
[17] Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness.,2005, 71(3): 1501–1506.
[18] Schloss PD, Handelsman J. Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures.,2006, 72(10): 6773–6779.
[19] Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data.,2010, 7(5): 335–336.
[20] Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo- Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS 2nd, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2.,2019, 37(8): 852–857.
[21] McDonald D, Vázquez-Baeza Y, Koslicki D, McClelland J, Reeve N, Xu Z, Gonzalez A, Knight R. Striped UniFrac: enabling microbiome analysis at unprecedented scale.,2018, 15(11): 847–848.
[22] Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Gregory Caporaso J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin.,2018, 6(1): 90.
[23] Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads., 17(1), doi: 10.14806/ej.17.1.200..
[24] Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data.,2016, 13(7): 581–583.
[25] Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics.,2016, 4: e2584.
[26] Bokulich NA, Dillon MR, Zhang Y, Rideout JR, Bolyen E, Li H, Albert PS, Caporaso JG. Q2-longitudinal: longitudinal and paired-sample analyses of microbiome data.,2018, 3(6): e00219–00218.
[27] Edgar RC. Search and clustering orders of magnitude faster than BLAST.,2010, 26(19): 2460–2461.
[28] Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection.,2011, 27(16): 2194–2200.
[29] Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads.,2013, 10(10): 996–998.
[30] Edgar RC, Flyvbjerg H. Error filtering, pair assembly and error correction for next-generation sequencing reads.,2015, 31(21): 3476–3482.
[31] Oksanen J, Kindt R, Legendre P, O’Hara B, Stevens MHH, Oksanen MJ, Suggests M. The vegan package.,2007, 10: 631–637.
[32] McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data.,2013, 8(4): e61217.
[33] Lahti L, Shetty S. Microbiome R package.,2012-2019. doi: 10.18129/B9.bioc.microbiome.
[34] McMurdie PJ, Holmes S. Shiny-phyloseq: web application for interactive microbiome analysis with provenance tracking.,2014, 31(2): 282–283.
[35] Gonzalez A, Navas-Molina JA, Kosciolek T, McDonald D, Vázquez-Baeza Y, Ackermann G, DeReus J, Janssen S, Swafford AD, Orchanian SB, Sanders JG, Shorenstein J, Holste H, Petrus S, Robbins-Pianka A, Brislawn CJ, Wang M, Rideout JR, Bolyen E, Dillon M, Caporaso JG, Dorrestein PC, Knight R. Qiita: rapid, web-enabled microbiome meta-analysis.,2018, 15(10): 796–798.
[36] Mitchell AL, Scheremetjew M, Denise H, Potter S, Tarkowska A, Qureshi M, Salazar GA, Pesseat S, Boland MA, Hunter FMI, Ten Hoopen P, Alako B, Amid C, Wilkinson DJ, Curtis TP, Cochrane G, Finn RD. EBI Metagenomics in 2017: enriching the analysis of microbial communities, from sequence reads to assemblies.,2018, 46(D1): D726–D735.
[37] Shi W, Qi H, Sun Q, Fan G, Liu S, Wang J, Zhu B, Liu H, Zhao F, Wang X, Hu X, Li W, Liu J, Tian Y, Wu L, Ma J. GcMeta: a global catalogue of metagenomics platform to support the archiving, standardization and analysis of microbiome data.,2018, 47(D1): D637–D648.
[38] McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea.,2012, 6(3): 610–618.
[39] Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools.,2013, 41 (Database issue): D590–596.
[40] Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. Ribosomal Database Project: data and tools for high throughput rRNA analysis.,2014, 42(Database issue): D633–D642.
[41] Nilsson RH, Larsson K-H, Taylor AFS, Bengtsson- Palme J, Jeppesen TS, Schigel D, Kennedy P, Picard K, Glöckner FO, Tedersoo L, Saar I, Kõljalg U, Abarenkov K. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications.,2019, 47(D1): D259–D264.
[42] Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences.,2013, 31(9): 814–821.
[43] Aßhauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data.,2015, 31(17): 2882– 2884.
[44] Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome.,2016, 353(6305): 1272–1277.
[45] Ward T, Larson J, Meulemans J, Hillmann B, Lynch J, Sidiropoulos D, Spear JR, Caporaso G, Blekhman R, Knight R, Fink R, Knights D. BugBase predicts organism-level microbiome phenotypes.,2017: 133462.
[46] Nguyen NH, Song Z, Bates ST, Branco S, Tedersoo L, Menke J, Schilling JS, Kennedy PG. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild.,2016, 20: 241–248.
[47] Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling.,2015, 12(10): 902–903.
[48] Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments.,2014, 15(3): R46.
[49] Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, Huttenhower C. Species-level functional profiling of metagenomes and metatranscriptomes.,2018, 15(11): 962–968.
[50] Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. MetaSPAdes: a new versatile metagenomic assembler.,2017, 27(5): 824–834.
[51] Seemann T. Prokka: rapid prokaryotic genome annotation.,2014, 30(14): 2068–2069.
[52] Lomsadze A, Gemayel K, Tang S, Borodovsky M. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes.,2018, 28(7): 1079–1089.
[53] Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data.,2012, 28(23): 3150–3152.
[54] Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013.,2014, 42(Database issue): D490–D495.
[55] Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN, Doshi S, Courtot M, Lo R, Williams LE, Frye JG, Elsayegh T, Sardar D, Westman EL, Pawlowski AC, Johnson TA, Brinkman FSL, Wright GD, McArthur AG. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database.,2017, 45(D1): D566–D573.
[56] Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface.,2019, 47(D1): D687– D692.
[57] Kang D, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies.,2019, 7: e7359.
[58] Wu YW, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets.,2015, 32(4): 605–607.
[59] Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C. Binning metagenomic contigs by coverage and composition.,2014, 11(11): 1144– 1146.
[60] Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis.,2018, 6(1): 158.
[61] Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, Banfield JF. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy.,2018, 3(7): 836–843.
[62] Ji P, Zhang Y, Wang J, Zhao F. MetaSort untangles metagenome assembly by reducing microbial community complexity.,2017, 8: 14306.
[63] Bishara A, Moss EL, Kolmogorov M, Parada AE, Weng Z, Sidow A, Dekas AE, Batzoglou S, Bhatt AS. High- quality genome sequences of uncultured microbes by assembly of read clouds.,2018, 36(11): 1067–1075.
[64] Bertrand D, Shaw J, Kalathiyappan M, Ng AHQ, Kumar MS, Li C, Dvornicic M, Soldo JP, Koh JY, Tong C, Ng OT, Barkham T, Young B, Marimuthu K, Chng KR, Sikic M, Nagarajan N. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes.,2019, 37(8): 937–944.
[65] Stewart RD, Auffret MD, Warr A, Walker AW, Roehe R, Watson M. Compendium of 4,941 rumen metagenome- assembled genomes for rumen microbiome biology and enzyme discovery.,2019, 37(8): 953– 961.
[66] Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report.,2016, 32(19): 3047– 3048.
[67] Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data.,2014, 30(15): 2114–2120.
[68] Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2.,2012, 9(4): 357–359.
[69] Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, UniProt Consortium. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches.,2015, 31(6): 926–932.
[70] Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph.,2015, 31(10): 1674–1676.
[71] Mikheenko A, Saveliev V, Gurevich A. MetaQUAST: evaluation of metagenome assemblies.,2016, 32(7): 1088–1090.
[72] Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression.,2017, 14(4): 417– 419.
[73] Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND.,2015, 12(1): 59–60.
[74] Huerta-Cepas J, Szklarczyk D, Heller D, Hernández- Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen Lars J, von Mering C, Bork P. EggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses.,2019, 47(D1): D309–D314.
[75] Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences.,2016, 428(4): 726–731.
[76] Gibson MK, Forsberg KJ, Dantas G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology.,2014, 9(1): 207–216.
[77] Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy.,2015, 16(1): 157.
[78] Comeau AM, Douglas GM, Langille MGI. Microbiome helper: a custom and streamlined workflow for microbiome research.,2017, 2(1): e00127–00116.
[79] Robinson MD, McCarthy DJ, Smyth GK. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data.,2010, 26(1): 139–140.
[80] Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.,2014, 15(12): 550.
[81] Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies.,2015, 43(7): e47.
[82] Bates D, Mächler M, Bolker B, Walker S. Fitting Linear Mixed-Effects Models using lme4., 2014.
[83] Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles.,2014, 30(21): 3123–3124.
[84] Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation.,2011, 12(6): R60.
[85] Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn.,2015, 3: e1029.
[86] Dhariwal A, Chong J, Habib S, King IL, Agellon LB, Xia J. MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data.,2017, 45(W1): W180–W188.
[87] Röttjers L, Faust K. From hairballs to hypotheses– biological insights from microbial networks.,2018, 42(6): 761–780.
[88] Banerjee S, Schlaeppi K, van der Heijden MGA. Keystone taxa as drivers of microbiome structure and functioning.,2018, 16(9): 567–576.
[89] Deng Y, Jiang YH, Yang Y, He Z, Luo F, Zhou J. Molecular ecological network analyses.,2012, 13(1): 113.
[90] Durno WE, Hanson NW, Konwar KM, Hallam SJ. Expanding the boundaries of local similarity analysis.,2013, 14(1): S3.
[91] Friedman J, Alm EJ. Inferring correlation networks from genomic survey data.,2012, 8(9): e1002687.
[92] Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks.,2003, 13(11): 2498–2504.
[93] Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C. relationships in the human microbiome.,2012, 8(7): e1002606.
[94] Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis.,2008, 9(1): 559.
[95] Kurtz ZD, Müller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Sparse and compositionally robust inference of microbial ecological networks.,2015, 11(5): e1004226.
[96] Csardi G, Nepusz T. The igraph software package for complex network research.,,2006, 1695(5): 1–9.
[97] Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. In Third international AAAI conference on weblogs and social media: 2009.
[98] Fan K, Weisenhorn P, Gilbert JA, Shi Y, Bai Y, Chu H. Soil pH correlates with the co-occurrence and assemblage process of diazotrophic communities in rhizosphere and bulk soils of wheat fields.,2018, 121: 185–192.
[99] Wang J, Zheng J, Shi W, Du N, Xu X, Zhang Y, Ji P, Zhang F, Jia Z, Wang Y, Zheng Z, Zhang H, Zhao F. Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus.,2018, 67(9): 1614–1625.
[100] Wang J, Jia Z, Zhang B, Peng L, Zhao F. Tracing the accumulation of in vivo human oral microbiota elucidates microbial community dynamics at the gateway to the GI tract.,2019: gutjnl-2019-318977.
[101] Katoh K, Standley DM. MAFFT Multiple sequence alignment software version 7: improvements in performance and usability.,2013, 30(4): 772–780.
[102] Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput.,2004, 32(5): 1792–1797.
[103] Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments.,2010, 5(3): e9490.
[104] Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies.,2015, 32(1): 268–274.
[105] Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis.,2016, 44(W1): W232–W235.
[106] Subramanian B, Gao S, Lercher MJ, Hu S, Chen WH. Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees.,2019, 47(W1): W270–W275.
[107] Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments.,2019, 47(W1): W256–W259.
[108] Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data.,2017, 8(1): 28–36.
[109] LeCun Y, Bengio Y, Hinton G. Deep learning.,2015, 521: 436–444.
[110] Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mähler A, Balogh A, Markó L, Vvedenskaya O, Kleiner FH, Tsvetkov D, Klug L, Costea PI, Sunagawa S, Maier L, Rakova N, Schatz V, Neubert P, Frätzer C, Krannich A, Gollasch M, Grohme DA, Côrte-Real BF, Gerlach RG, Basic M, Typas A, Wu C, Titze JM, Jantsch J, Boschmann M, Dechend R, Kleinewietfeld M, Kempa S, Bork P, Linker RA, Alm EJ, Müller DN. Salt-responsive gut commensal modulates TH17 axis and disease.,2017, 551(7682): 585–589.
[111] Ren Z, Li A, Jiang J, Zhou L, Yu Z, Lu H, Xie H, Chen X, Shao L, Zhang R, Xu S, Zhang H, Cui G, Chen X, Sun R, Wen H, Lerut JP, Kan Q, Li L, Zheng S. Gut microbiome analysis as a tool towards targeted non- invasive biomarkers for early hepatocellular carcinoma.,2019, 68(6): 1014–1023.
[112] Metcalf JL, Xu ZZ, Weiss S, Lax S, van Treuren W, Hyde ER, Song SJ, Amir A, Larsen P, Sangwan N, Haarmann D, Humphrey GC, Ackermann G, Thompson LR, Lauber C, Bibat A, Nicholas C, Gebert MJ, Petrosino JF, Reed SC, Gilbert JA, Lynne AM, Bucheli SR, Carter DO, Knight R. Microbial community assembly and metabolic function during mammalian corpse decomposition.,2016, 351(6269): 158–162.
[113] Zhang J, Zhang N, Liu YX, Zhang X, Hu B, Qin Y, Xu H, Wang H, Guo X, Qian J, Wang W, Zhang P, Jin T, Chu C, Bai Y. Root microbiota shift in rice correlates with resident time in the field and developmental stage.,2018, 61(6): 613–621.
[114] Liaw A, Wiener M. Classification and regression by randomForest.,2002, 2(3): 18–22.
[115] Galkin F, Aliper A, Putin E, Kuznetsov I, Gladyshev VN, Zhavoronkov A. Human microbiome aging clocks based on deep learning and tandem of permutation feature importance and accumulated local effects.,2018, 507780.
[116] Yang C, Yang RF, Cui YJ. Bacterial genome-wide association study: methodologies and applications., 2018, 40(1): 57–65.杨超, 杨瑞馥, 崔玉军. 细菌全基因组关联研究的方法与应用. 遗传,2018, 40(1): 57–65.
[117] Wang J, Thingholm LB, Skiecevičienė J, Rausch P, Kummen M, Hov JR, Degenhardt F, Heinsen FA, Rühlemann MC, Szymczak S, Holm K, Esko T, Sun J, Pricop-Jeckstadt M, Al-Dury S, Bohov P, Bethune J, Sommer F, Ellinghaus D, Berge RK, Hübenthal M, Koch M, Schwarz K, Rimbach G, Hübbe P, Pan WH, Sheibani-Tezerji R, Häsler R, Rosenstiel P, D'Amato M, Cloppenborg-Schmidt K, Künzel S, Laudes M, Marschall HU, Lieb W, Nöthlings U, Karlsen TH, Baines JF, Franke A. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota.,2016, 48(11): 1396–1406.
[118] Wang J, Chen L, Zhao N, Xu X, Xu Y, Zhu B. Of genes and microbes: solving the intricacies in host genomes.,2018, 9(5): 446–461.
[119] Jin T, Wang Y, Huang Y, Xu J, Zhang P, Wang N, Liu X, Chu H, Liu G, Jiang H, Li Y, Xu J, Kristiansen K, Xiao L, Zhang Y, Zhang G, Du G, Zhang H, Zou H, Zhang H, Jie Z, Liang S, Jia H, Wan J, Lin D, Li J, Fan G, Yang H, Wang J, Bai Y, Xu X. Taxonomic structure and functional association of foxtail millet root microbiome.,2017, 6(10): 1–12.
[120] Wang Z, Lu G, Yuan M, Yu H, Wang S, Li X, Deng Y. Elevated temperature overrides the effects of N amendment in Tibetan grassland on soil microbiome.,2019, 136: 107532.
[121] Shi Y, Li Y, Xiang X, Sun R, Yang T, He D, Zhang K, Ni Y, Zhu YG, Adams JM, Chu H. Spatial scale affects the relative role of stochasticity versus determinism in soil bacterial communities in wheat fields across the North China Plain.,2018, 6(1): 27.
[122] Zhang K, Shi Y, Cui X, Yue P, Li K, Liu X, Tripathi BM, Chu H. Salinity is a key determinant for soil microbial communities in a desert ecosystem.,2019, 4(1): e00225–00218.
[123] Doherty MK, Ding T, Koumpouras C, Telesco SE, Monast C, Das A, Brodmerkel C, Schloss PD. Fecal microbiota signatures are associated with response to ustekinumab therapy among crohn’s disease patients.,2018, 9(2): e02120–02117.
[124] DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, Sun CL, Goltsman DSA, Wong RJ, Shaw G, Stevenson DK, Holmes SP, Relman DA. Temporal and spatial variation of the human microbiota during pregnancy.,2015, 112(35): 11060–11065.
[125] Garrido-Oter R, Nakano RT, Dombrowski N, Ma KW, McHardy AC, Schulze-Lefert P. Modular traits of the Rhizobiales root microbiota and their evolutionary relationship with symbiotic Rhizobia.,2018, 24(1): 155–167.e5.
[126] Castrillo G, Teixeira PL, Paredes SH, Law TF, de Lorenzo L, Feltcher ME, Finkel OM, Breakfield NW, Mieczkowski P, Jones CD, Paz-Ares J, Dangl JL. Root microbiota drive direct integration of phosphate stress and immunity.,2017, 543(7646): 513–518.
[127] Herrera Paredes S, Gao T, Law TF, Finkel OM, Mucyn T, Teixeira PJPL, Salas González I, Feltcher ME, Powers MJ, Shank EA, Jones CD, Jojic V, Dangl JL, Castrillo G. Design of synthetic bacterial communities for predictable plant phenotypes.,2018, 16(2): e2003962.
[128] Almeida A, Mitchell AL, Boland M, Forster SC, Gloor GB, Tarkowska A, Lawley TD, Finn RD. A new genomic blueprint of the human gut microbiota.,2019, 568(7753): 499–504.
[129] Vandeputte D, Kathagen G, D’hoe K, Vieira-Silva S, Valles-Colomer M, Sabino J, Wang J, Tito RY, De Commer L, Darzi Y, Vermeire S, Falony G, Raes J. Quantitative microbiome profiling links gut community variation to microbial load.,2017, 551(7681): 507–511.
[130] Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, Ross MC, Lloyd RE, Doddapaneni H, Metcalf GA, Muzny D, Gibbs RA, Vatanen T, Huttenhower C, Xavier RJ, Rewers M, Hagopian W, Toppari J, Ziegler AG, She JX, Akolkar B, Lernmark A, Hyoty H, Vehik K, Krischer JP, Petrosino JF. Temporal development of the gut microbiome in early childhood from the TEDDY study.,2018, 562(7728): 583–588.
[131] Meadow JF, Altrichter AE, Kembel SW, Moriyama M, O’Connor TK, Womack AM, Brown GZ, Green JL, Bohannan BJM. Bacterial communities on classroom surfaces vary with human contact.,2014, 2(1): 7.
[132] Huang AC, Jiang T, Liu YX, Bai YC, Reed J, Qu B, Goossens A, Nützmann HW, Bai Y, Osbourn A. A specialized metabolic network selectively modulatesroot microbiota.,2019, 364(6440): eaau6389.
[133] Chen Q, Jiang T, Liu YX, Liu H, Zhao T, Liu Z, Gan X, Hallab A, Wang X, He J, Ma Y, Zhang F, Jin T, Schranz ME, Wang Y, Bai Y, Wang G. Recently duplicated sesterterpene (C25) gene clusters inthaliana modulate root microbiota.,2019, 62(7): 947–958.
[134] Charalampous T, Kay GL, Richardson H, Aydin A, Baldan R, Jeanes C, Rae D, Grundy S, Turner DJ, Wain J, Leggett RM, Livermore DM, O’Grady J. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection.,2019, 37(7): 783–792.
[135] Bradley P, den Bakker HC, Rocha EPC, McVean G, Iqbal Z. Ultrafast search of all deposited bacterial and viral genomic data.,2019, 37(2): 152– 159.
[136] Pasolli E, Schiffer L, Manghi P, Renson A, Obenchain V, Truong DT, Beghini F, Malik F, Ramos M, Dowd JB, Huttenhower C, Morgan M, Segata N, Waldron L. Accessible, curated metagenomic data through.,2017, 14(11): 1023–1024.
[137] Vangay P, Hillmann BM, Knights D. Microbiome learning repo (ML Repo): a public repository of microbiome regression and classification tasks.,2019, 8(5).
[138] Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, Collado MC, Rice BL, DuLong C, Morgan XC, Golden CD, Quince C, Huttenhower C, Segata N. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle.,2019, 176(3): 649–662.e20.
Methods and applications for microbiome data analysis
Yongxin Liu1,2, Yuan Qin1,2,3, Xiaoxuan Guo1,2, Yang Bai1,2,3
Development of high-throughput sequencing stimulates a series of microbiome technologies, such as amplicon sequencing, metagenome, metatranscriptome, which have rapidly promoted microbiome research. Microbiome data analysis involves a lot of basic knowledge, software and databases, and it is difficult for peers to learn and select proper methods. This review systematically outlines the basic ideas of microbiome data analysis and the basic knowledge required to conduct analysis. In addition, it summarizes the advantages and disadvantages of commonly used software and databases used in the comparison, visualization, network, evolution, machine learning and association analysis. This review aims to provide a convenient and flexible guide for selecting analytical tools and suitable databases for mining the biological significance of microbiome data.
microbiome; data analysis; amplicon; metagenome; pipeline
2019-07-30;
2019-08-21
中国科学院前沿科学重点研究项目(编号:QYZDB-SSW-SMC021),国家自然科学基金面上项目(编号:31772400)和中国科学院重点部署项目(编号:KFZD-SW-219)资助[Supported by the Key Research Program of Frontier Sciences of the Chinese Academy of Science (No. QYZDB-SSW-SMC021), the National Natural Science Foundation of China (No. 31772400), and the Key Research Program of the Chinese Academy of Sciences (No. KFZD-SW-219)]
刘永鑫,博士,工程师,研究方向:生物信息学、宏基因组学。E-mail: yxliu@genetics.ac.cn
白洋,博士,研究员,研究方向:根系微生物组。E-mail: ybai@genetics.ac.cn
10.16288/j.yczz.19-222
2019/9/2 16:21:23
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190902.1620.001.html
(责任编委: 赵方庆)
