Gitelman综合征合并干燥综合征1例病例报告
2019-09-23王斌刘爱云郭秀燕王怀荣
王斌 刘爱云 郭秀燕 王怀荣
【摘要】在临床上,低钾血症非常常见,而Gitelman综合征是一种非常少见的低钾血症类型,临床表现为低钾血症、低镁血症、低尿钙、血气分析显示为代谢性碱中毒。在1966年由Gitelman首次发现并报道,由于SLC12A3基因突变导致Na+重吸收减少,从而继发性肾素激活,醛固酮增多,排钾增多最终导致低血钾。文献报道的Gitelman综合征的发病率为四万分之一,医师根据临床症状判断常有缺陷,近年随着基因检测技术的进步,越来越多的Gitelman综合征被诊断。本文收治并通过基因诊断一例Gitelman综合征合并干燥综合征患者,报告如下。
【关键词】Gitelman综合征;低钾血症;SLC12A3基因
【中图分类号】R714.2 【文献标识码】A 【文章编号】1004-597X(2019)17-0003-02
【病历资料】患者女26岁,因“发热、四肢酸痛、乏力半月”于2018年2月4日入院。半月前开始发热,最高38.5℃,四肢酸痛、乏力,无咳嗽、咳痰、鼻塞、流涕,无晨僵、小关节肿痛,无心悸、盗汗,自服“布洛芬1片/次”,于2小时内体温降至正常。2018年2月1日门诊查肝功能正常,二氧化碳结合力43mmol/L,钙2.43mmol/L,钾1.59mmol/L,钠133mmol/L,氯86mmol/L,磷0.61mmol/L,镁0.95mmol/L,CRP 21.7mg/L,给予“氯化钾缓释片”口服,未规律服药症状加重,以“低钾血症”于2018年2月4日收入内分泌科。既往体健。生育2女体健。父母及1弟健康。体格检查:体温36.7℃,血压126/83mmhg,营养中等,双眼无突出,双侧甲状腺无肿大,心、肺、腹查体无明显异常,神经系统查体阴性。化验室检查:血分析:白细胞11.98×109/L,中性粒细胞10.27×109/L,红细胞4.30X1012/L,血红蛋白118g/L,血小板231×109/L。ALT、AST正常,总蛋白92g/L,球蛋白52.4g/L,白蛋白44.5g/L,白球比0.85g/L,二氧化碳结合力43mmol/L,钙2.43mmol/L,钾1.59mmol/L,钠133mmol/L,氯86(99-110)mmol/L,磷0.61mmol/L,镁0.95mmol/L,同步24h尿钾50.97mmol/L,24h尿钠187.2mmol/L,24h尿氯180.8mmol/L,24h尿钙0.49mmol/L,随机尿钾48.14mmol/L,随机尿尿钾/肌酐:5.94mmol/mmol,血气分析:PH 7.65,PCO247mmhg,HCO3-51.8mmol/L,肌酸激酶205U/L,肌酸激酶同工酶205U/L,乳酸脱氢酶295U/L,a羟丁酸227U/L,血浆渗透压276mOsm/k,尿渗透压476mosm/k,血浆。肾素浓度(立位)301.6(4-38)pg/ml,醛固酮(立位)106.7(40-310)pg/ml,醛固酮/肾素浓度0.35(0-25),血管紧张素Ⅱ(立位)130.7(49-252)pg/ml。血浆肾素浓度(卧位)140.8(4-24)pg/ml,醛固酮(卧位)150.9(10-160)pg/ml,醛固酮/肾素浓度1.07(0-25),血管紧张素Ⅱ(卧位)128.9(25-129)pg/ml。血皮質醇483.3mmol/L,ACTH 17.58pg/ml,血沉104mm/h。入院后口服缓释片1.0g tid,静脉补钾6g,乳酸左氧氟沙星氯化钠0.5g iv qd控制感染,2018年2月5日复查电解质钾2.40mmol/L。胸片未见明显异常,尿分析+尿沉渣:WBC,RBC,亚硝酸盐,细菌数500(0-4000)/ul,(2018年2月7日)血沉90mm/h,风湿系列16项:抗核抗体:核颗粒型,抗核抗体滴度1:3200,抗SSA抗体+++,抗SSB抗体+,抗Ro-52抗体+++,免疫固定电泳M蛋白阴性,抗中性粒细胞浆抗体阴性,抗内皮细胞抗体IgG阴性,抗肾小球基底膜抗体阴性,抗蛋白酶3抗体阴性,抗髓过氧化物酶抗体阴性,抗心磷脂抗体IgA/G/M弱阳性,抗β2-糖蛋白1抗体IgG阴性,直接抗人球蛋白实验阴性,狼疮抗凝物检测1:32.8S,狼疮抗凝物检测233.8(30-38)S,抗凝物检测比率0.97。人类白细胞抗原-H27阴性。IgG22.47g/L,IgA 3.98g/L,IgM 1.34g/L,补体:C3:1.31g/L,C4:0.31g/L,AS0:304 IU/ml,直接抗人球蛋白实验阴性。抗心磷脂抗体IgA/G/M弱阳性,抗β2-糖蛋白1抗体IgG阴性。诊断为干燥综合征,停抗生素,给予羟氯喹200mg bid、强的松10mgqd、白芍总苷0.6g bid。2018年2月9日采集抗凝管血3ml,送至济南金域医学检验中心,肾脏穿刺活检:免疫荧光检查:共查到3个肾小球,未见明显荧光分布,IgG-、IgA-、IgM-、C3-、F-、Clq-、Kappa-、Lambda-。HE及特殊染色:共查见5个。肾小球,显示1个肾小球完全纤维化,肾小球系膜区无增殖,系膜细胞1-3个/系膜区,系膜基质无扩大,肾小球毛细血管壁无增厚,未见明显球旁器结构。Masson染色显示未见明显嗜复红蛋白沉积。肾小管上皮细胞肿胀,颗粒变性,可见个别蛋白管型,肾间质无明显纤维化,灶性淋巴细胞、单核细胞浸润,间质内小血管壁无增厚。无新月体。
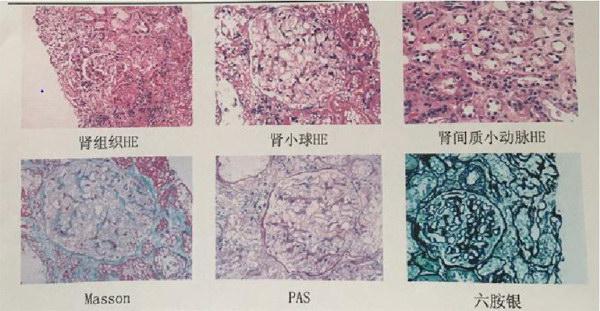
图1 北京市不同类别《尘肺病报告卡》从确诊至报告的时间间隔分布情况
2018年3月15日基因结果:16q13携带SLC12A3基因两个杂合致病变异,第一个错义突变是位于12号外显子(Exonl2),eDNA第1456位碱基由鸟嘌呤(G)突变为腺嘌呤(A),预计会使所编码蛋白质的第486位氨基酸由天冬氨酸(Asp)变为天冬酰胺(Asn),第二个错义突变是位于15号外显子(Exon15),eDNA第1924位碱基由胞嘧啶(C)突变为胸腺嘧啶(T),预计会使所编码蛋白质的第642位氨基酸由精氨酸(Arg)变为半胱氨酸(Cys)。随访,电解质钾3.05mmol/L、血钠140mmol/L、血氯100mmol/L、血沉52mm/h。
【讨论】Gitelman综合征是一种常染色体隐性遗传的失盐性。肾小管疾病,多以低血钙、低血镁、低尿钙及代谢性碱中毒为临床特点。患病率为四万分之一,青少年或成年人中均可发病。目前明确病因是由于编码位于肾远曲小管的噻嗪类利尿剂敏感的钠氯共同转运体(NCCT)蛋白的基因SLCl2A3发生功能缺失突变,从而导致NCCT的结构和/或功能异常,继而引起肾脏远曲小管对钠氯重吸收障碍,最终导致低血容量、肾素/血管紧张素一醛固酮系统(RAAS)的激活,以及低血钾和代谢性碱中毒。Gitelman综合征和Batter综合征临床表现比较相似,均可出现低血钾及。肾素激活,但二者的发病机制不同。以往基因检测技术不常应用于临床,Gitelman综合征被认为是Batter综合征的一个亚型,直至1966年Gitelman的基因被成功克隆。随诊基因检测技术的进步,Gitelman综合征被独立出来。钠离子和氯离子被肾小球滤过后,10%在肾远曲小管和集合管被重吸收,由编码基因位于染色体16q13的SLC12A3的钠氯共同转运体NCCT和编码基因位于染色体1p36的CLCNKB和远曲小管上皮细胞基膜侧钠钾ATP酶(Na-K-TPase)共同协作完成。当NCCT功能缺陷时,钠离子重吸收减少,镁离子通道(TRPM6)数量下降,镁离子重吸收减少,所以出现低血镁。镁的排泄主要依靠上皮TRPM6基因调控,在Gitelman综合征患者中由于基因突變导致尿镁排出增加从而引发低血镁,另外肾素激活、醛固酮的作用使得管腔侧钠离子重吸收增加,通过Na+/Mg2+交换增加,导致尿镁增多,血镁降低。低镁血症常被认为是Gitelman和Batter综合征的主要鉴别点,本患者血镁水平处在正常范围,随着基因诊断金标准的确立,血镁正常的Gitelman综合征更多的引起了关注,文献报道血镁正常的患者占8%-22%。传统意义上说,肾小球球旁器增生是Gitelman综合征的病理生理学基础,但并非100%发生,。肾穿刺活检并不是Gitelman确立的金标准,而有赖于基因诊断。
本患者特殊之处在于同时合并干燥综合征。其抗核抗体滴度1:3200,特异性抗SSA和抗SSB抗体阳性,干燥综合征诊断明确。干燥综合征{SS}是一种主要累及外分泌腺体的慢性炎症性自身免疫病。临床除有涎腺和泪腺分泌受损功能外,还可导致皮肤、骨骼肌肉、肾脏、肺脏、消化系统、神经系统等多系统损害。干燥综合征也可出现低血钾,主要原因是由于大量淋巴细胞浸润引起。肾小管上皮细胞退行性变,使得远端肾小管泌氢功能障碍,出现低血钾,尿液不能酸化,则出现碱性尿。
目前GS仍无治愈方法,治疗以对症支持来控制,单纯补钾治疗效果补不佳,需同时应用保钾利尿剂如螺内酯及阿米洛利,同时补镁。本病患者补钾治疗后血钾仍不能维持在正常水平,不排除随着干燥综合征的进展进一步加重肾小管损伤。随着病史延长,有必要重新进行。肾穿刺活检,进一步明确肾损害的程度,知道下一步治疗,改善预后。
