意大利蜜蜂响应东方蜜蜂微孢子虫胁迫的免疫应答
2019-09-23付中民陈华枝刘思亚祝智威范小雪范元婵万洁琦张璐熊翠玲徐国钧陈大福郭睿
付中民,陈华枝,刘思亚,祝智威,范小雪,范元婵,万洁琦,张璐,熊翠玲,徐国钧,陈大福,郭睿
意大利蜜蜂响应东方蜜蜂微孢子虫胁迫的免疫应答
付中民,陈华枝,刘思亚,祝智威,范小雪,范元婵,万洁琦,张璐,熊翠玲,徐国钧,陈大福,郭睿
(福建农林大学蜂学学院,福州 350002)
【】通过对意大利蜜蜂(,简称意蜂)工蜂中肠响应东方蜜蜂微孢子虫()胁迫的差异表达基因(differentially expressed gene,DEG)及免疫通路进行深入细致的分析,揭示宿主的细胞和体液免疫应答,为关键免疫应答基因的筛选及功能研究打下基础。基于前期获得的正常及东方蜜蜂微孢子虫胁迫的意蜂7日龄和10日龄工蜂中肠(Am7CK、Am7T、Am10CK、Am10T)转录组数据,按照FDR≤1,≤0.05和|log2fold change|≥1的标准筛选出各组的DEG,利用相关生物信息学软件对DEG进行Pearson相关性分析、Venn分析、GO分类和KEGG代谢通路富集分析,进而对免疫通路富集基因进行统计和分析,利用实时荧光定量PCR(real-time quantitative PCR,RT-qPCR)验证转录组数据的可靠性。差异分析结果显示,Am7CK vs Am7T比较组包含472个上调基因和385个下调基因;Am10CK vs Am10T比较组包含611个上调基因和360个下调基因。Venn分析结果显示,两个比较组特有的DEG分别为739和853个,共有的DEG为118个。GO分类结果显示,Am7CK vs Am7T比较组中上调和下调基因分别涉及23和29个功能条目,其中富集上调基因数最多的前5位分别为结合、催化活性、代谢进程、细胞进程和单组织进程;富集下调基因数最多的前5位分别为代谢进程、单组织进程、催化活性、细胞进程和结合。Am10CK vs Am10T比较组中上调和下调基因分别涉及36和26个功能条目,其中富集上调基因数最多的前5位分别为单组织进程、结合、细胞进程、催化活性和代谢活性;富集下调基因数最多的前5位分别为结合、细胞进程、催化活性、代谢进程和单组织进程。KEGG代谢通路富集分析结果显示,Am7CK vs Am7T比较组中上调和下调基因分别富集在38和33条代谢通路,富集上调基因数最多的前5条分别为胆汁分泌、内质网蛋白加工、泛素介导的蛋白水解、PI3K-Akt信号通路和神经营养因子信号通路;富集下调基因数最多的前5条分别为胞质DNA传感通路、嘌呤代谢、嘧啶代谢、RNA聚合酶和核糖体;涉及泛素介导的蛋白水解等3条细胞免疫通路,以及PI3K-Akt信号通路等7条体液免疫通路。Am10CK vs Am10T比较组中上调和下调基因分别富集在54和43条代谢通路,富集上调基因数最多的前5条分别为Hippo信号通路、药物代谢-细胞色素P450、P450对外源物质的代谢、泛素介导的蛋白水解和鞘脂类代谢;富集下调基因数最多的前5条分别为mRNA监测、鞘脂类信号通路、果糖和甘露糖代谢、半乳糖代谢和鞘脂类代谢;涉及泛素介导的蛋白水解等7条细胞免疫通路,以及NF-B信号通路等2条体液免疫通路。RT-qPCR验证结果显示6个随机挑选的DEG的表达量变化趋势与测序结果一致,证实了本研究中测序数据的可靠性。进一步分析发现意蜂7日龄和10日龄工蜂中肠的NF-B信号通路均被东方蜜蜂微孢子虫激活,随即启动了3种抗菌肽apidaecin、defensin-1和hymenoptaecin的合成,体现了它们在宿主抵御东方蜜蜂微孢子虫入侵中的重要性。在转录组水平解析了意蜂工蜂中肠对东方蜜蜂微孢子虫的免疫应答,揭示宿主在胁迫前期同时作出细胞和体液免疫应答,前者可能在抵御病原入侵方面发挥主要作用;宿主的细胞免疫在胁迫后期持续增强,但体液免疫较大程度地减弱;泛素介导的蛋白水解通路及富集DEG,NF-B信号通路及富集DEG,以及抗菌肽编码基因、和可能在意蜂工蜂对东方蜜蜂微孢子虫的免疫应答中起到关键作用。
意大利蜜蜂;东方蜜蜂微孢子虫;中肠;免疫应答;细胞免疫;体液免疫
0 引言
【研究意义】蜜蜂是一种专食植物蜜粉的昆虫,可为70%农作物和大部分野生植物授粉[1],对农业生产、生态环境和生物多样性至关重要[2]。意大利蜜蜂(,简称意蜂)属于西方蜜蜂(),为我国饲养规模最大的蜂种,经济效益显著[3]。东方蜜蜂微孢子虫()特异性地侵染成年蜜蜂中肠上皮细胞,对蜂王、工蜂和雄蜂均有侵染性[4-5],可对蜜蜂造成诸多方面的影响,如细胞破坏[6]、能量胁迫[7-8]和寿命缩短[9]等,还能与其他生物或非生物胁迫因子共同危害蜂群[10-11]。近年来,欧洲和美国等地的西方蜜蜂相继暴发蜂群崩溃综合症,即蜂群中只剩下蜂王、卵及一些未成年的工蜂和大量蜜粉残留于巢脾内,大量的成年工蜂短期内突然在巢外消失,该病已被证明与东方蜜蜂微孢子虫密切相关[12-14]。目前,关于蜜蜂响应东方蜜蜂微孢子虫的免疫应答机制尚不明确。利用最新的组学技术对意蜂工蜂中肠响应东方蜜蜂微孢子虫胁迫的差异表达基因(differentially expressed gene,DEG)及其潜在功能进行系统分析,可在组学水平揭示意蜂对东方蜜蜂微孢子虫的免疫应答,为在分子水平阐明宿主的免疫应答机制提供重要依据。【前人研究进展】前人对东方蜜蜂微孢子虫的研究主要集中在流行病学、致病机理及诊断防治[15-17]等方面。Cornman等于2009年测序、组装并注释了东方蜜蜂微孢子虫的参考基因组[18],为其组学及分子生物学研究奠定了基础。此后,在意蜂对东方蜜蜂微孢子虫的胁迫应答方面进行了一些组学研究,但相关信息总体仍较为有限[19-20]。Badaoui等通过RNA-seq技术研究了东方蜜蜂微孢子虫感染后5、10和15 d西方蜜蜂的基因表达情况,发现宿主的抗菌肽、角质层蛋白和气味结合蛋白编码基因表达量均为下调,导致宿主的免疫功能下降,从而促进微孢子虫在蜂群中的存活和增殖[19];Aufauvre等利用二代测序和实时荧光定量PCR(real-time quantitative PCR,RT-qPCR)技术对东方蜜蜂微孢子虫单独胁迫、杀虫剂单独胁迫及二者共同胁迫1 d和7 d的西方蜜蜂中肠样品进行研究,结果显示东方蜜蜂微孢子虫与杀虫剂共同胁迫虽然导致西方蜜蜂的死亡率显著上升,但二者不存在协同效应;此外,无论是东方蜜蜂微孢子虫单独胁迫还是与杀虫剂共同胁迫,都能导致宿主中肠免疫相关基因、角质层基因和海藻糖代谢相关基因表达量的显著改变[20]。笔者团队前期已利用RNA-seq技术对正常及东方蜜蜂微孢子虫胁迫的意蜂7 d和10 d工蜂中肠进行了全转录组测序,在高质量组学数据的基础上对高表达基因(highly expressed gene,HEG)表达谱进行了初步分析[21];全面解析了意蜂工蜂中肠发育过程中长链非编码RNA(long non-coding RNA,lncRNA)和环状RNA(circular RNA,circRNA)的差异表达谱、调控网络及潜在作用机制[22-23]。上述研究结果为本研究提供了较好的数据支持和前期基础。【本研究切入点】前期研究中,笔者团队在mRNA组学水平解析了意蜂10日龄工蜂中肠响应东方蜜蜂微孢子虫胁迫的HEG表达谱及HEG的潜在作用,为揭示宿主的胁迫应答机制提供了一些有益信息。但仅从基因表达量角度进行分析具有一定的局限性。DEG与东方蜜蜂微孢子虫胁迫存在直接的联系,因此,本研究在前期分析的基础上,进一步从基因表达差异变化倍数的角度对意蜂工蜂中肠响应东方蜜蜂微孢子虫胁迫的DEG表达谱及免疫应答进行分析和探讨。【拟解决的关键问题】基于前期获得的高质量全转录组数据[21-23],对意蜂工蜂中肠响应东方蜜蜂微孢子虫胁迫的DEG表达谱及潜在功能进行细致的生物信息学分析和分子生物学验证,进而对宿主的细胞和体液免疫通路及富集DEG进行深入分析,在转录组层面揭示意蜂对东方蜜蜂微孢子虫胁迫的免疫应答。
1 材料与方法
供试意蜂工蜂取自福建农林大学蜂学学院教学蜂场。东方蜜蜂微孢子虫孢子由福建农林大学蜂学学院蜜蜂保护实验室纯化和保存。试验于2017年在福建农林大学完成。
1.1 意大利蜜蜂工蜂中肠全转录组数据来源
笔者所在课题组前期已利用Illumina技术对正常及东方蜜蜂微孢子虫胁迫的意蜂工蜂中肠进行全转录组测序,通过严格的质控和过滤获得了高质量的测序数据[22-23]。
工蜂中肠的制备及测序简述如下:将刚出房的工蜂放入灭菌的四周打孔的塑料盒(20只/盒),每个盒子上方插一支装有50%(w/v)无菌糖水的饲喂器。将工蜂出房当天记为0 d,(34±0.5)℃培养24 h。饥饿处理2 h,单头固定,处理组工蜂每头饲喂含纯化孢子(1×106个)的糖水5 μL,对照组工蜂每头饲喂不含孢子的糖水5 μL,24 h后均饲喂不含孢子的糖水,每日更换糖水,及时清理未食尽糖水的工蜂及死去的工蜂。饲喂至7 d和10 d,分别拉取中肠,置于RNA-Free的EP管,放入液氮速冻。然后迅速转移到-80℃超低温冰箱保存备用。每次取样时间均控制在20 s内。处理组和对照组均包含3个生物学重复。意蜂7日龄工蜂中肠对照组和处理组样品为Am7CK:Am7CK1、Am7CK2、Am7CK3;Am7T:Am7T1、Am7T2、Am7T3;意蜂10日龄工蜂中肠对照组和处理组样品为Am10CK:Am10CK1、Am10CK2、Am10CK3;Am10T:Am10T1、Am10T2、Am10T3。参照郭睿和陈大福等[22,24]的方法对上述12个中肠样品进行RNA抽提和cDNA文库构建,委托广州基迪奥生物技术有限公司进行双端测序,测序平台为Illumina HiSeq 4000。
1.2 测序数据质控、差异表达基因(DEG)筛选及生物信息学分析
利用短reads比对工具bowtie将前期测序得到的各中肠样品的有效读段(clean reads)映射到核糖体数据库,去除比对上核糖体的reads后,将剩余的reads映射到西方蜜蜂参考基因组(Aml_4.5),比对上的clean reads用于后续的生物信息学分析。
利用公式FPKM=Total exon fragment/[Mapped fragment (millions)×exon length (kB)]计算所有表达基因的FPKM值。利用R软件计算各样品之间的相关性系数。DEG的筛选标准为FDR≤1,≤0.05和|log2fold change|≥1。利用在线分析工具集合OmicsShare(http://www.omicshare.com/)进行GO分类和KEGG代谢通路富集分析等相关生物信息学分析,采用默认参数。
1.3 DEG的实时荧光定量PCR(RT-qPCR)验证
为了验证全转录组测序数据的可靠性,随机选取6个DEG(TCONS_00035591、TCONS_00021864、XM_006569665.2、TCONS_00030754、XM_003251768.3和XM_016912533.1)进行RT-qPCR验证。参照郭睿等[21]的方法,利用DNAMAN软件根据所选DEG的碱基序列设计相应的特异性引物,委托上海生工生物工程有限公司进行合成,相关引物信息详见表1。利用RNA抽提试剂盒(TaKaRa公司,中国)分别提取Am7CK、Am7T、Am10CK和Am10T样品的总RNA,以Oligo (dT)23作为引物进行反转录,得到对应的cDNA,作为DEG和内参基因的qPCR模板。反应体系(20 μL)包含上下游向引物(10.0 μmol·L-1)各1 μL,cDNA模板DNA 1 μL,SYBR Green Dye 10 μL,DEPC水7 μL。反应在QuantStudio荧光定量PCR仪(ThermoFisher公司,美国)上进行,按照SYBR Green Dye试剂盒(Vazyme公司,中国)操作说明书上的方法,每个反应进行3次重复。反应条件:95℃预变性1 min,95℃变性15 s,60℃退火30 s,共40个循环,最后72℃延伸45 s。利用2-ΔΔCt法对上述DEG的相对表达量进行计算。通过GraphPad Prism软件进行相关数据分析及绘图。

表1 RT-qPCR验证使用的引物信息
2 结果
2.1 比对西方蜜蜂参考基因组数据分析
前期研究结果表明Am7CK和Am10CK的组内各样品相关性均在0.95以上[23]。Am7T的组内各样品相关性介于0.976—1.000,Am10T的组内各样品相关性均介于0.977—1.000(图1),表明处理组的样品重复性较好。通过映射核糖体数据库去除rRNA的reads,共得到1 668 076 502条clean reads,进而通过映射西方蜜蜂基因组得到505 845 865条能够比对上的clean reads,Q30均在95.16%以上,说明测序数据质量良好,可用于后续分析。
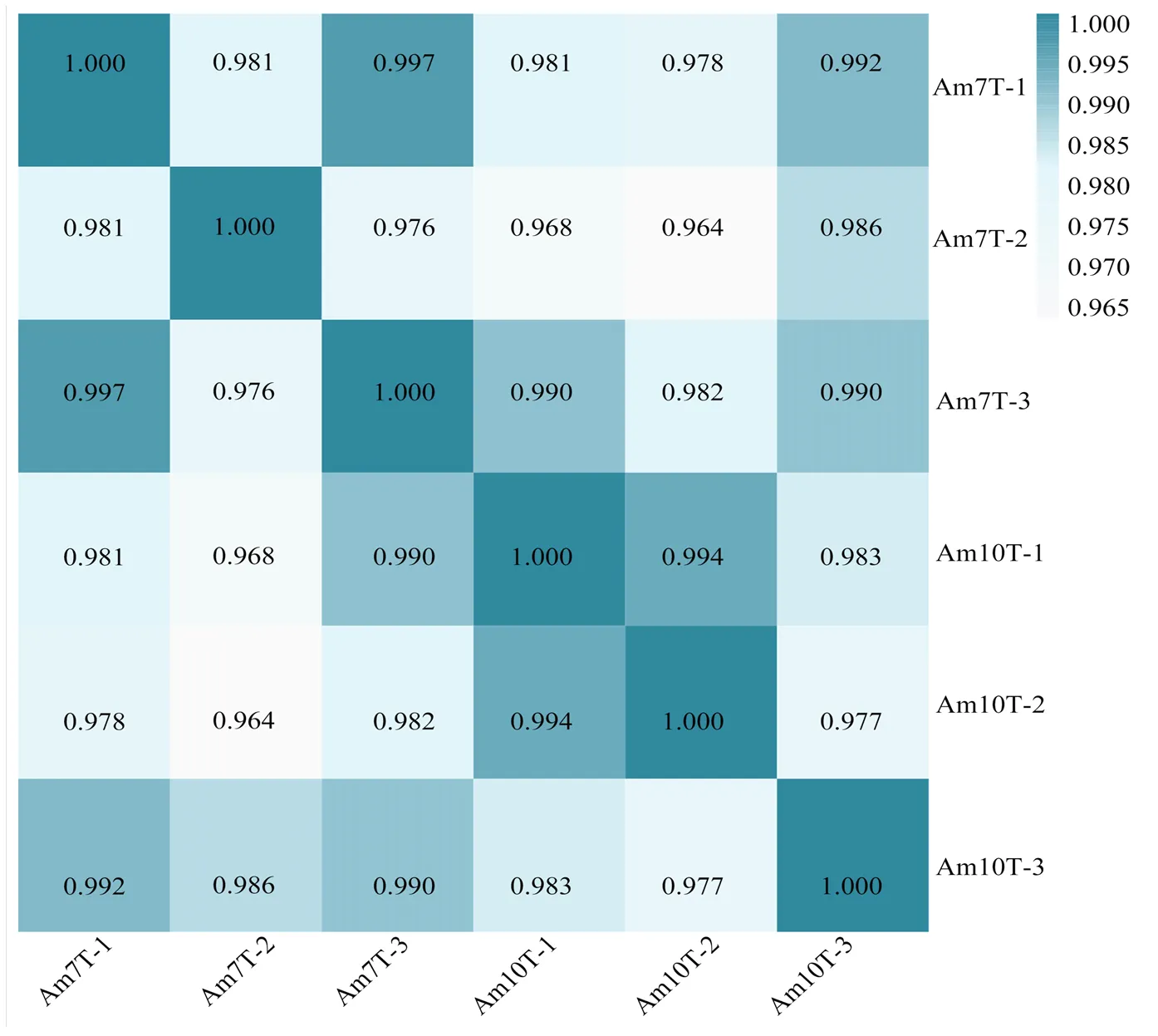
图1 各东方蜜蜂微孢子虫感染的意蜂工蜂中肠样品组内不同生物学重复间的Pearson相关性
2.2 差异表达基因分析
差异分析结果显示,Am7CK vs Am7T比较组中共有857个基因差异表达,其中上调基因为472个,下调基因为385个(图2-A);Am10CK vs Am10T比较组包含971个DEG,其中上调和下调基因数分别为611和360个(图2-B)。进一步对上述两个比较组的DEG进行Venn分析,结果显示二者特有DEG分别为739和853个,共有DEG为118个(图2-C)。
2.3 DEG的GO分类
GO分类结果显示,Am7CK vs Am7T中的DEG主要分为生物学进程、细胞组分和分子功能3类,其中上调基因涉及23个功能条目,富集基因数最多的前5位分别是结合(26)、催化活性(23)、代谢进程(17)、细胞进程(17)和单组织进程(14);下调基因分布在29个功能条目,富集基因数最多的前5位分别是代谢进程(12)、单组织进程(11)、催化活性(11)、细胞进程(10)和结合(9)(图3-A—C)。
Am10CK vs Am10T中的DEG同样分为生物学进程、细胞组分和分子功能,其中上调基因涉及36个功能条目,富集基因数最多的前5位分别是单组织进程(27)、结合(27)、细胞进程(25)、催化活性(23)和代谢进程(20);下调基因分布在26个功能条目,富集基因数最多的前5位分别是结合(23)、细胞进程(22)、催化活性(21)、代谢进程(21)和单组织进程(17)(图3-D—E)。
2.4 DEG的KEGG代谢通路富集分析
代谢通路富集分析结果显示,Am7CK vs Am7T中上调基因涉及38条代谢通路,富集基因数最多的分别是胆汁分泌(2)、内质网蛋白加工(2)、泛素介导的蛋白水解(2)、PI3K-Akt信号通路(2)、神经营养因子信号通路(1)、趋化因子信号通路(1)、近端小管碳酸氢盐再生(1)、集管酸分泌(1)、催乳激素分泌(1)和胃酸分泌(1);下调基因涉及33条代谢通路,富集基因数最多的分别是胞质DNA传感通路(2)、嘌呤代谢(2)、嘧啶代谢(2)、RNA聚合酶(2)、核糖体(2)、脂肪的消化和吸收(1)、血管平滑肌收缩(1)、蛋白质的消化和吸收(1)、胰液分泌(1)和氧化磷酸化(1)。

A:Am7CK vs Am7T中DEG的火山图Volcano plot of DEGs in Am7CK vs Am7T;B:Am10CK vs Am10T中DEG的火山图Volcano plot of DEGs in Am10CK vs Am10T。红色圆点代表上调基因,绿色圆点代表下调基因Red dots indicate up-regulated genes, green dots indicate down-regulated genes。C:Am7CK vs Am7T和Am10CK vs Am10T中DEG的Venn图 Venn diagram of DEGs in Am7CK vs Am7T and Am10CK vs Am10T
Am10CK vs Am10T中的上调基因涉及54条代谢通路,富集基因数最多的分别是Hippo信号通路(3)、药物代谢-细胞色素P450(2)、P450对外源物质的代谢(2)、泛素介导的蛋白水解(2)、鞘脂类代谢(2)、氨基糖和核苷酸代谢(2)、药物代谢-其他酶(2)、鞘脂类信号通路(2)、Hedgehog信号通路(2)和血小板激活(1);下调基因涉及43条代谢通路,富集基因数最多的分别是mRNA监控(3)、鞘脂类信号通路(3)、果糖和甘露糖代谢(2)、半乳糖代谢(2)、鞘脂类代谢(2)、氨基糖和核苷酸糖代谢(2)、RNA转运(2)、心肌细胞的肾上腺素能信号(1)、突出囊泡循环(1)和背腹轴形成(1)。
2.5 细胞和体液免疫通路分析
进一步对免疫通路及富集基因进行分析,发现Am7CK vs Am7T中的DEG可注释到3条细胞免疫通路,包括泛素介导的蛋白水解、细胞凋亡和溶酶体(表2);以及7条体液免疫通路,包括PI3K-Akt信号通路、趋化因子信号通路、NF-B信号通路、cAMP信号通路、Ras信号通路、MAPK信号通路-酵母和FoxO信号通路(表3)。对于细胞免疫通路,富集在泛素介导的蛋白水解信号通路上的2个基因均上调表达,富集在细胞凋亡上的1个基因也呈现出表达上调,但富集在溶酶体上的1个基因表达量下调。对于体液免疫通路,富集在趋化因子信号通路和NF-B信号通路上的基因均为上调表达,但富集在cAMP信号通路、Ras信号通路、MAPK信号通路-酵母和FoxO信号通路上的基因均表现为下调。

A—C:Am7CK vs Am7T中上调和下调基因分别在生物学进程、分子功能和细胞组分相关条目的分类Classifications of up- and down-regulated genes within Am7CK vs Am7T comparison group in GO terms associated with biological process, molecular function and cellular component;D—F:Am10CK vs Am10T中上调和下调基因分别在生物学进程、分子功能和细胞组分相关条目的分类Classifications of up- and down-regulated genes within Am10CK vs Am10T comparison group in GO terms associated with biological process, molecular function and cellular component
在Am10CK vs Am10T中,DEG分别注释到7条细胞免疫通路和2条体液免疫通路。对于细胞免疫通路,富集在泛素介导的蛋白水解、P450对外源物质代谢、药物代谢-细胞色素P450通路、细胞凋亡、Fc--R介导的吞噬作用和溶酶体通路上的基因都呈现出上调表达,仅有1个富集在内吞作用上的基因表现为下调(表4)。对于体液免疫通路,分别有1个上调基因和1个下调基因富集在NF-B信号通路和PI3K-Akt信号通路(表5)。
Am7CK vs Am7T和Am10CK vs Am10T比较组中DEG在泛素介导的蛋白水解上的富集详情如图4所示。

表2 Am7CK vs Am7T中DEG富集的细胞免疫相关通路
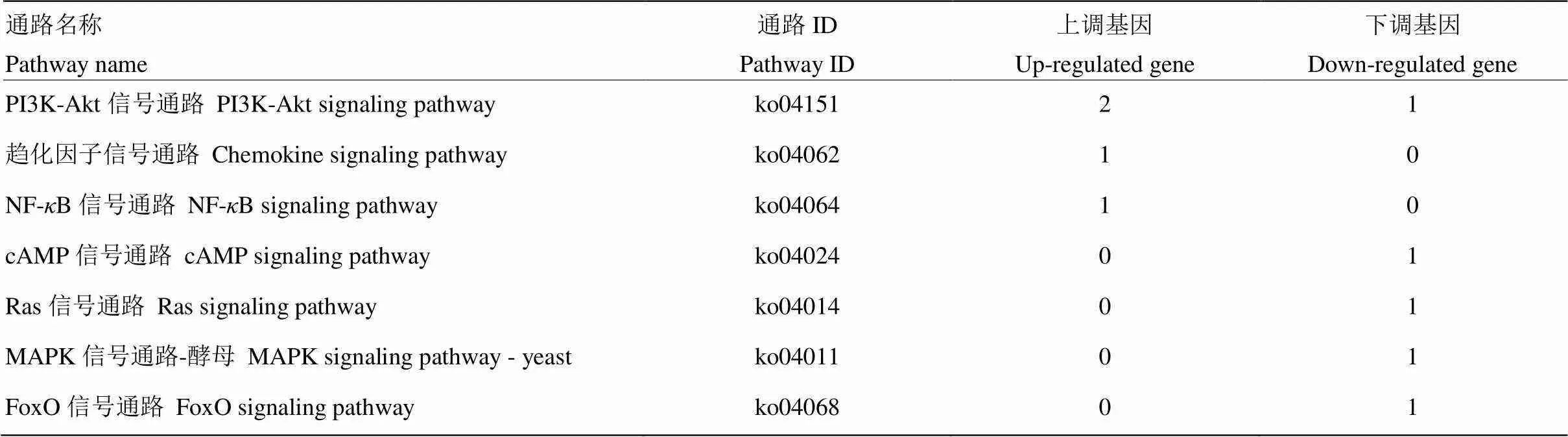
表3 Am7CK vs Am7T中DEG富集的体液免疫相关通路

表4 Am10CK vs Am10T中DEG富集的细胞免疫相关通路

表5 Am10CK vs Am10T中DEG富集的体液免疫相关通路

A:Am7CK vs Am7T中DEG在泛素介导蛋白水解上的富集情况Enrichment of DEGs within Am7CK vs Am7T comparison group in ubiquitin mediated proteolysis;B:Am10CK vs Am10T中DEG在泛素介导蛋白水解上的富集情况Enrichment of DEGs within Am10CK vs Am10T comparison group in ubiquitin mediated proteolysis。红色方框代表上调基因Red boxes represent up-regulated genes
2.6 DEG的RT-qPCR验证
RT-qPCR检测结果显示,6个DEG的表达水平变化趋势与测序数据中的基因表达水平变化趋势一致(图5),证明本研究中的mRNA组学数据真实可靠。

A:乙酰转移酶编码基因(TCONS_00035591)predicted acetyltransferase encoded gene;B:牙本质涎磷蛋白编码基因(TCONS_00021864)dentin sialophosphoprotein encoded gene;C:富含甘氨酸的细胞壁结构蛋白1.8-like型X2编码基因(XM_006569665.2) glycine-rich cell wall structural protein 1.8-like isoform X2 encoded gene;D:金星激酶受体编码基因(TCONS_00030754)venus kinase receptor encoded gene;E:G12蛋白编码基因(XM_003251768.3) protein G12 encoded gene;F:内切型2编码基因(XM_016912533.1)endochitinase isoform 2encoded gene。RT-qPCR组中,*表示p<0.05,**表示p<0.01 In RT-qPCR group, * indicates p<0.05, ** indicates p<0.01
3 讨论
前人对于蜜蜂响应东方蜜蜂微孢子虫的胁迫应答虽有一些研究[18-20],但多集中在分子生物学水平,组学相关研究较为有限。前期研究中,笔者团队利用RNA-seq技术对正常及东方蜜蜂微孢子虫胁迫的意蜂工蜂中肠进行了全转录组数据,获得了高质量的mRNA、miRNA、lncRNA和circRNA组学数据[21-23];通过生物信息学方法筛选出对照组和处理组的高表达基因(HEG),并通过深入分析揭示了HEG的表达谱及潜在作用[21]。HEG分析是从基因表达量角度对宿主的胁迫应答进行初步分析,虽然能得到一些有益信息,但具有不可避免的局限性,体现在有些基因对于意蜂工蜂中肠的生长发育十分重要,其表达水平自始至终都维持高量表达,这部分HEG会对相关分析造成一些影响;此外,HEG的分析只涉及被东方蜜蜂微孢子虫激活的基因,但被抑制的基因同样值得深入研究。免疫应答是一个复杂的动态过程,阐明其背后的分子机制需要从不同角度尽可能深入地进行分析,以获取尽可能多的有效信息。与HEG相比,差异表达基因(DEG)与东方蜜蜂微孢子虫胁迫存在直接的联系。为进一步探讨意蜂工蜂中肠响应东方蜜蜂微孢子虫胁迫的免疫应答,本研究在前期研究基础上对DEG及其潜在作用进行相关分析,发现Am7CK vs Am7T中的上调和下调基因数分别为472和385个;Am10CK vs Am10T中的上调和下调基因数分别为611和360个;表明随着胁迫时间的延长,更多的宿主基因被激活,而受到抑制的基因数基本一致,胁迫应答水平呈增强趋势。进一步分析发现,有118个DEG为上述两个比较组所共有,特有DEG的数量分别为739和853个,推测这些共有DEG在宿主的胁迫应答过程发挥基础性的作用,而特有DEG在宿主胁迫应答的不同阶段具有特定功能。此外,将Am10CK vs Am10T中的DEG与前期筛选出的Am10T的HEG[21]进行Venn分析,结果显示有5个基因既为高量表达又为差异表达,包括半胱氨酸调节子的转录调节剂(transcriptional regulator for cysteine regulon)编码基因、蜂毒肽前体蛋白(melittin precursor)编码基因、白细胞蛋白酶抑制剂(antileukoproteinase-like isoform X2)编码基因、硫氧还原蛋白(thioredoxin-2 isoform 1)编码基因和胰蛋白酶(trypsin-7)编码基因,推测这些基因在意蜂工蜂响应东方蜜蜂微孢子虫胁迫的过程中发挥关键作用,值得进一步深入研究。下一步将针对上述5个基因进行全长序列的分子克隆及功能研究(例如RNAi敲减),以在分子水平探究它们的功能。
富集在细胞杀伤和应激反应条目的DEG可能与免疫应答密切相关。本研究发现,Am7CK vs Am7T中分别有6个上调基因和4个下调基因富集在应激反应,2个下调基因富集在细胞杀伤;Am10CK vs Am10T中分别有3个上调基因和3个下调基因富集在应激反应,1个上调基因和1个下调基因富集在细胞杀伤;表明部分基因处于激活状态、部分基因处于抑制状态,暗示着意蜂工蜂在对东方蜜蜂微孢子虫的胁迫响应过程,既能产生直接的免疫应答,也会受到病原一定程度的抑制,反映出宿主和病原之间存在复杂的互作关系。
蜜蜂的免疫防御分为群体免疫和个体免疫,后者又分为细胞免疫和体液免疫[25]。本研究中,在东方蜜蜂微孢子虫胁迫的意蜂7日龄工蜂中肠中,分别有2个(log2fold change=1.766和1.685)和1个上调基因(log2fold change=1.685)富集在泛素介导的蛋白水解和细胞凋亡,仅有1个下调基因(log2fold change= -1.02)富集在溶酶体;分别有1个(log2fold change=12.805)和1个上调基因(log2fold change=1.685)富集在趋化因子和NF-B信号通路,1个(log2fold change=-9.103)、1个(log2fold change=-1.938)、1个(log2fold change= -13.391)和1个下调基因(log2fold change=-13.391)富集在cAMP、Ras、MAPK和FoxO信号通路;分别有2个上调基因(log2fold change=12.805和1.766)和1个下调基因(log2fold change=-2.023)富集在PI3K-Akt;表明意蜂工蜂中肠的细胞免疫很大程度被激活,而体液免疫部分程度被抑制、部分程度被激活。在东方蜜蜂微孢子虫胁迫的意蜂10日龄工蜂中肠中,富集在细胞免疫通路的绝大多数DEG都为上调表达,仅有1个富集在内吞作用的基因下调表达(log2fold change=-1.543);分别有1个上调基因(log2fold change= 1.016)和1个下调基因(log2fold change= -2.569)富集在NF-B和PI3K-Akt信号通路;表明随着东方蜜蜂微孢子虫胁迫时间的延长,意蜂工蜂中肠的细胞免疫被持续激活且激活程度不断增强,而体液免疫的种类减少、程度下降。上述结果说明意蜂工蜂中肠在东方蜜蜂微孢子虫胁迫的前期,免疫系统识别到病原入侵,同时作出细胞和体液应答,此时宿主的细胞免疫很大程度被激活,在抵御病原入侵方面发挥主要作用,宿主的体液免疫既有部分被激活也有部分被抑制,体现了意蜂及东方蜜蜂微孢子虫互作的复杂性;在东方蜜蜂微孢子虫胁迫的后期,宿主的细胞免疫更大范围、更大程度地被激活,在免疫防御中的作用继续增强,而宿主的体液免疫通路较大程度地下降,其中NF-B信号通路呈现出激活状态,PI3K-Akt信号通路呈现出抑制状态。
泛素介导的蛋白水解是一种经典的细胞免疫通路,该通路由E1酶(泛素活化酶)、E2酶(泛素结合酶)和E3酶(泛素连接酶)组成,其中E3可特异性识别不同底物,然后与E2酶结合,E2酶集合E1酶活化的泛素,泛素与底物形成多聚泛素链,最终被蛋白质酶体识别降解[26]。本研究中,对于东方蜜蜂微孢子虫胁迫的意蜂7日龄和10日龄工蜂中肠,共有4个上调基因编码3种E3酶,推测宿主通过提高E3酶的合成以识别东方蜜蜂微孢子虫的关键蛋白质,从而对其产生抑制。另外,病原也能够利用泛素介导的蛋白水解的不同环节以逃避宿主细胞免疫系统的监控[27]。蜜蜂白垩病是另一种常见的蜜蜂真菌病,该病的病原蜜蜂球囊菌(,简称球囊菌)特异性侵染蜜蜂幼虫,早期的增殖场所为幼虫肠道。前期研究中,笔者团队发现在球囊菌胁迫的中华蜜蜂()幼虫肠道中,靶向结合泛素介导的蛋白水解通路上的3个基因的miRNA-21-x表达量显著上调,作者推测球囊菌通过调控宿主泛素介导的蛋白水解通路基因的表达以协助球囊菌逃避宿主免疫系统的监控[28]。东方蜜蜂微孢子虫特异性寄生蜜蜂中肠上皮细胞,其能否利用miRNA介导的方法调控泛素介导的蛋白水解通路基因从而逃脱宿主的免疫系统监控,值得进一步深入研究。陈阳研究发现泛素介导的蛋白水解与Toll样受体(TLRs)信号通路共同参与鸭的基因介导的抗病毒天然免疫信号的传递途径[29]。果蝇的Toll免疫信号通路主要识别革兰氏阳性菌和真菌等病原微生物,通过激活Toll受体等一系列信号分子激活NF-B同源蛋白Dif和Dorsal并释放到核内,最终启动下游抗菌肽分子的表达[30]。本研究发现,意蜂7日龄和10日龄工蜂中肠的NF-B信号通路都被激活,且启动了抗菌肽编码基因、和(NM_001011613.1、NM_001011615.1和NM_001011616.2)的表达,表明NF-B信号通路在宿主免疫应答中表现活跃。综上,推测泛素介导的蛋白水解通路及富集DEG,NF-B信号通路及富集DEG,以及、和在意蜂工蜂对东方蜜蜂微孢子虫的免疫应答中发挥关键作用。
关于mRNA与非编码RNA(non-coding RNA,ncRNA)之间的相互作用,Salmena等[31]提出了“竞争性内源RNA”假说,即任何包含miRNA反应元件(miRNA response element)的RNA如mRNA、假基因、lncRNA和circRNA,均可竞争性地结合miRNA,从而影响基因的表达水平。前期研究中,笔者团队基于高质量的全转录组数据对意蜂工蜂中肠发育过程的ncRNA进行了系统的差异表达谱及调控网络分析,筛选出若干与中肠发育密切相关的关键lncRNA和circRNA[22-23]。本研究对意蜂工蜂中肠响应东方蜜蜂微孢子虫胁迫的DEG进行了全面分析,得到了宿主响应东方蜜蜂微孢子虫胁迫的基因表达谱信息,但相关基因集仍较为复杂。目前,笔者团队正在对意蜂工蜂响应东方蜜蜂微孢子虫胁迫的差异表达miRNA、差异表达lncRNA和差异表达circRNA进行深入分析,拟通过构建和分析DEG与差异表达ncRNA之间的ceRNA调控网络,筛选出居于核心位置的关键应答基因,作为后续功能研究的分子靶标。
4 结论
在mRNA组学水平对意蜂工蜂中肠响应东方蜜蜂微孢子虫胁迫的免疫应答进行了深入解析,发现意蜂工蜂中肠在胁迫前期同时作出细胞和体液免疫应答,前者可能在抵御病原入侵方面发挥主要作用;宿主的细胞免疫在胁迫后期的作用持续增强,但体液免疫的作用较大程度的减弱;泛素介导的蛋白水解通路及富集DEG,NF-B信号通路及富集DEG,以及、和在意蜂工蜂对东方蜜蜂微孢子虫的免疫应答中起到关键作用。研究结果为在分子水平阐明意蜂响应东方蜜蜂微孢子虫的免疫应答机制提供了数据支持。
[1] KLEIN A M, VAISSIÈRE B E, CANE J H, STEFFAN-DEWENTER I, CUNNINGHAM S A, KREMEN C, TSCHARNTKE T. Importance of pollinators in changing landscapes for world crops., 2007, 274(1608): 303-313.
[2] POTTS S G, BIESMEIJER J C, KREMEN C, NEUMANN P, SCHWEIGER O, KUNIN W E. Global pollinator declines: trends, impacts and drivers., 2010, 25(6): 345-353.
[3] 曾志将, 颜伟玉, 饶波, 谢宪斌. 蜜蜂性比研究进展. 上海交通大学学报(农业科学版), 2003, 21(2): 164-167.
ZENG Z J, YAN W Y, RAO B, XIE X B. Advances on the honeybee sex ratio., 2003, 21(2): 164-167. (in Chinese)
[4] TRAVER B E, FELL R D. Low natural levels ofinqueens., 2012, 110(3): 408-410.
[5] TRAVER B E, FELL R D.in drone honey bees ()., 2011, 107(3): 234-236.
[6] GARCIA-PALENCIA P, MARTIN-HERNANDEZ R, GONZALEZ- PORTO A V, MARIN P, MEANA A, HIGES M. Natural infection bycauses similar lesions as in experimentally infected caged-worker honey bees ()., 2010, 49(3): 278-283.
[7] MAYACK C, NAUG D. Energetic stress in the honeybeefrominfection., 2009, 100(3): 185-188.
[8] MARTIN-HERNANDEZ R, BOTIAS C, BARRIOS L, MARTINEZ- SALVADOR A, MEANA A, MAYACK C, HIGES M. Comparison of the energetic stress associated with experimentalandinfection of honeybees ()., 2011, 109(3): 605-612.
[9] LECOCQ A, JENSEN A B, KRYGER P, NIEH J C. Parasite infection accelerates age polyethism in young honey bees., 2016, 6: 22042.
[10] SHUTLER D, HEAD K, BURGHER-MACLELLAN K L, COLWELL M J, LEVITT A L, OSTIGUY N, WILLIAMS G R. Honey beeparasites in the absence offungi andmites., 2014, 9(6): e98599.
[11] WU J Y, SMART M D, ANELLI C M, SHEPPARD W S. Honey bees () reared in brood combs containing high levels of pesticide residues exhibit increased susceptibility to(Microsporidia) infection., 2012, 109(3): 326-329.
[12] COX-FOSTER D L, CONLAN S, HOLMES E C, PALACIOS G, EVANS J D, MORAN N A, QUAN P L, BRIESE T, HORNIG M, GEISER D M, MARTINSON V, VANENGELSDORP D, KALKSTEIN A L, DRYSDALE A, HUI J, ZHAI J, CUI L, HUTCHISON S K, SIMONS J F, EGHOLM M, PETTIS J S, LIPKIN W I. A metagenomic survey of microbes in honey bee colony collapse disorder., 2007, 318(5848): 283-287.
[13] CHEJANOVSKY N, OPHIR R, SCHWAGER M S, SLABEZKI Y, GROSSMAN S, COX-FOSTER D. Characterization of viral siRNA populations in honey bee colony collapse disorder., 2014, 454/455: 176-183.
[14] KRIBS-ZALETA C M, MITCHELL C. Modeling colony collapse disorder in honeybees as a contagion., 2014, 11(6): 1275-1294.
[15] MAYACK C, NATSOPOULOU M E, MCMAHON D P.alters a highly conserved hormonal stress pathway in honeybees., 2015, 24(6): 662-670.
[16] NATSOPOULOU M E, DOUBLET V, PAXTON R J. European isolates of the microsporidiaandhave similar virulence in laboratory tests on European worker honey bees., 2016, 47(1): 57-65.
[17] BRAVO J, CARBONELL V, SEPÚLVEDA B, DELPORTE C, VALDOVINOS C E, MARTÍN-HERNÁNDEZ R, HIGES M. Antifungal activity of the essential oil obtained fromagainst infection in honey bees by., 2017, 149: 141-147.
[18] CORNMAN R S, CHEN Y P, SCHATZ M C, STREET C, ZHAO Y, DESANY B, EGHOLM M, HUTCHISON S, PETTIS J S, LIPKIN W I, EVANS J D. Genomic analyses of the microsporidian, an emergent pathogen of honey bees., 2009, 5(6): e1000466.
[19] BADAOUI B, FOUGEROUX A, PETIT F, ANSELMO A, GORNI C, CUCURACHI M, CERSINI A, GRANATO A, CARDETI G, FORMATO G, MUTINELLI F, GIUFFRA E, WILLIAMS J L, BOTTI S. RNA-sequence analysis of gene expression from honeybees ()infected with., 2017, 12(3): e0173438.
[20] AUFAUVRE J, MISME-AUCOUTURIER B, Viques B, TEXIER C, DELBAC F, BLOT N. Transcriptome analyses of the honeybee response toand insecticides., 2014, 9(3): e91686.
[21] 郭睿, 刀晨, 熊翠玲, 郑燕珍, 付中民, 耿四海, 陈大福. 意大利蜜蜂工蜂中肠响应胁迫的高表达基因分析. 环境昆虫学报, 2018, 40(5): 1106-1112.
GUO R, DAO C, XIONG C L, ZHENG Y Z, FU Z M, GENG S H, CHEN D F. Analysis of the highly expressed genes in the midgut ofworker under the stress of., 2018, 40(5): 1106-1112. (in Chinese)
[22] 郭睿, 耿四海, 熊翠玲, 郑燕珍, 付中民, 王海朋, 杜宇, 童新宇, 赵红霞, 陈大福. 意大利蜜蜂工蜂中肠发育过程中长链非编码RNA的差异表达分析. 中国农业科学, 2018, 51(18): 3600-3613.
GUO R, GENG S H, XIONG C L, ZHENG Y Z, FU Z M, WANG H P, DU Y, TONG X Y, ZHAO H X, CHEN D F. Differential expression analysis of long non-coding RNAs during the developmental process ofworker’s midgut., 2018, 51(18): 3600-3613. (in Chinese)
[23] 郭睿, 陈华枝, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 杜宇, 王海朋, 耿四海, 周丁丁, 刘思亚, 陈大福. 意大利蜜蜂工蜂中肠发育过程中的差异表达环状RNA及其调控网络分析. 中国农业科学, 2018, 51(23): 4575-4590.
GUO R, CHEN H Z, XIONG C L, ZHENG Y Z, FU Z M, XU G J, DU Y, WANG H P, GENG S H, ZHOU D D, LIU S Y, CHEN D FAnalysis of differentially expressed circular RNAs and their regulation networks during the developmental process ofworker’s midgut, 2018, 51(23): 4575-4590. (in Chinese)
[24] 陈大福, 郭睿, 熊翠玲, 梁勤, 郑燕珍, 徐细建, 张曌楠, 黄枳腱, 张璐, 王鸿权, 解彦玲, 童新宇. 中华蜜蜂幼虫肠道响应球囊菌早期胁迫的转录组学. 中国农业科学, 2017, 50(13): 2614-2623.
CHEN D F, GUO R, XIONG C L, LIANG Q, ZHENg Y Z, XU X J, ZHANG Z N, HUANG Z J, ZHANG L, WANG H Q, XIE Y L, TONG X Y. Transcriptome oflarval gut under the stress of., 2017, 50(13): 2614-2623. (in Chinese)
[25] ARONSTEIN K A, MURRAY K D. Chalkbrood disease in honey bees., 2010, 103(Suppl. 1): S20-S29.
[26] CIECHANOVER A, 葛亮. 泛素介导的蛋白质降解系统——从基础研究到临床应用. 生命科学, 2010, 22(3): 212-215.
CIECHANOVER A, GE L. The ubiquitin proteolytic system—from bench to the bedside., 2010, 22(3): 212-215. (in Chinese)
[27] 李朝飞, 庞义. 泛素-蛋白水解酶复合体通路与病毒侵染. 生物工程学报, 2004, 20(2): 151-156.
LI C F, PANG Y. Ubiquitin-proteasome pathway and virus infection., 2004, 20(2): 151-156. (in Chinese)
[28] 杜宇, 童新宇, 周丁丁, 陈大福, 熊翠玲, 郑燕珍, 徐国钧, 王海朋, 陈华枝, 郭意龙, 隆琦, 郭睿. 中华蜜蜂幼虫肠道响应球囊菌胁迫的microRNA应答分析. 微生物学报, 2019. DOI: 10.13343/j.cnki. wsxb.20180401.
DU Y, TONG X Y, ZHOU D D, CHEN D F, XIONG C L, ZHENG Y Z, XU G J, WANG H P, CHEN H Z, GUO Y L, LONG Q, GUO R. MicroRNA responses in the larval gut oftostress., 2019. DOI: 10. 13343/j.cnki.wsxb.20180401. (in Chinese)
[29] 陈阳. 鸭RIG-I在免疫调节中的作用及分子调控机制[D]. 扬州: 扬州大学, 2015.
CHEN Y. Function and molecular mechanism of duck RIG-I gene in immune regulation[D]. Yangzhou: Yangzhou University, 2015. (in Chinese)
[30] 徐娇. miR-964在果蝇Toll免疫响应中的调控作用研究[D]. 南京: 南京师范大学, 2018.
XU J. Study on the regulation of miR-964 in the immune response of drosophila Toll[D]. Nanjing: Nanjing Normal University, 2018. (in Chinese)
[31] SALMENA L, POLISENO L, TAY Y, KATS L, PANDOLFI P P. A ceRNA hypothesis: the rosetta stone of a hidden RNA language?, 2011, 146(3): 353-358.
Immune responses oftostress
FU zhongMin, CHEN HuaZhi, LIU SiYa, ZHU ZhiWei, FAN XiaoXue, FAN YuanChan, WAN JieQi, ZHANG Lu, XIONG CuiLing, XU GuoJun, CHEN DaFu, GUO Rui
(College of Bee Science, Fujian Agriculture and Forestry University, Fuzhou 350002)
【】The objective of this study is to reveal the cellular and humoral immune responses ofworker’s midgut tostress, and to provide a basis for screening and functional study of key immune response genes by deep investigation of host differentially expressed genes (DEGs) and immune pathways.【】Base on the previously obtained transcriptome datasets of normal and-stressed midguts of7- and 10-day-old workers’ midgut samples (Am7CK, Am7T, Am10CK and Am10T), DEGs in each comparison group were filtered out following the standard FDR≤1,≤0.05 and |log2fold change|≥1. Additionally, Pearson correlations, Venn analysis, GO classification and KEGG pathway enrichment analysis were conducted by using related bioinformatic softwares. Further, DEGs enriched in immune pathways were summarized and analyzed. Finally, the reliability of transcriptome data was verified via real-time quantitative PCR (RT-qPCR).【】Differential expression analysis showed that there were 472 up-regulated and 385 down-regulated genes in Am7CK vs Am7T comparison group, and 611 up-regulated and 360 down-regulated genes in Am10CK vs Am10T comparison group. Venn analysis suggested that the number of specific DEGs of the aforementioned two comparison groups was 739 and 853, respectively, and 118 DEGs were shared. GO classification indicated that the up- and down-regulated genes in Am7CK vs Am7T were respectively annotated to 23 and 29 functional terms; among them, the top five terms of up-regulated genes were binding, catalytic activity, metabolic process, cellular process and single tissue process; while the top five terms of down-regulated genes were metabolic process, single tissue process, catalytic activity, cellular process and binding. In addition, the up- and down-regulated genes in Am10CK vs Am10T were respectively involved in 36 and 26 functional terms; among them, the top five terms of up-regulated genes were single process, binding, cellular process, catalytic activity and metabolic activity; while the top five terms of down-regulated genes were binding, cellular process, catalytic activity, metabolic process and single process. KEGG metabolic pathway enrichment analysis showed that the up- and down-regulated genes in Am7CK vs Am7T were respectively enriched in 38 and 33 pathways, and among them the top five enriched pathways of up-regulated genes were bile secretion, endoplasmic reticulum protein processing, ubiquitin-mediated proteolysis, PI3K-Akt signaling pathway and neurotrophic factor signaling pathway; while the top five enriched pathways of down-regulated genes were cytoplasmic DNA-sensing pathway, purine metabolism, pyrimidine metabolism, RNA polymerase and ribosome; three cellular immune pathways including ubiquitin-mediated proteolysis, and seven humoral pathways including PI3K-Akt signaling pathway were detected. Additionally, the up- and down-regulated genes in Am10CK vs Am10T were respectively associated with 54 and 43 pathways, and among them the top five enriched pathways of up-regulated genes were Hippo signaling pathway, drug metabolism-cytochrome P450, metabolism of xenobiotics by cytochrome P450, ubiquitin-mediated proteolysis, and sphingolipid metabolism; while the top five enriched pathways of down-regulated genes were mRNA surveillance pathway, sphingolipid signaling pathway, fructose and mannose metabolism, galactose metabolism, and sphingolipid metabolism; seven cellular immune pathways including ubiquitin-mediated proteolysis, and two humoral pathways including NF-B signaling pathway were found. The result of RT-qPCR verification demonstrated the expression trend of six randomly selected DEGs was consistent with that of the sequencing data, which confirmed the reliability of the transcriptome data. Further analysis noted that NF-B signaling pathway was activated byin both 7- and 10-day-old workers’ midguts of, followed by immediate initiation of three antimicrobial peptides such as apidaecin, defensin-1 and hymenoptaecin, indicating their key importance in host defense againstinvasion.【】The immune responses ofworker’s midgut towere uncovered at transcriptomic level, and it was revealed that the host made both cellular and humoral immune responses at the early stage ofstress. the host cellular immune responses may play a major role in resisting pathogen invasion. The host cellular immune continued to increase at the later stage of fungal stress, while the humoral immune drastically weakened. The ubiquitin mediated proteolysis and enriched DEGs, NF-B signaling pathway and enriched DEGs, and antibacterial peptide-encoded genes such as,andwere likely to play pivotal roles in host immune response tostress.
;; midgut; immune response; cellular immune; humoral immune

2019-04-17;
2019-05-24
国家现代农业产业技术体系建设专项资金(CARS-44-KXJ7)、福建省自然科学基金(2018J05042)、福建省教育厅中青年教师教育科研项目(JAT170158)、福建农林大学杰出青年科研人才计划(xjq201814)、福建农林大学科技创新专项基金(CXZX2017342,CXZX2017343)、福建省大学生创新创业训练计划(3165602032,3155006018)
付中民,E-mail:369699776@qq.com。陈华枝,E-mail:18965015689@163.com。付中民和陈华枝为同等贡献作者。通信作者郭睿,E-mail:ruiguo@fafu.edu.cn
(责任编辑 岳梅)
