顶空-气相色谱-质谱法同时测定7种甲磺酸酯类杂质
2019-09-23王少敏刘宏民
王少敏, 牛 颖, 代 敏, 魏 一, 刘宏民
(1.郑州大学 化学与分子工程学院 河南 郑州 450001; 2. 新药创制与药物安全性评价河南省协同创新中心 河南 郑州 450001)
0 引言
基因毒性物质是一类能对DNA结构造成破坏而产生致癌性的物质,因此对药物中此类物质的检测引起人们广泛关注[1-3].在制药过程中,烷基磺酸常用作原料药的成盐试剂来提高药物的稳定性和溶解性,磺酸酯的前体物质(如磺酰氯)也常用作反应试剂或催化剂,这些应用都可能生成磺酸酯类基因毒性物质[4].替比培南酯是第1个可口服的碳青霉烯类抗生素,对大多数临床分离的菌株均具有比青霉素及头孢类抗生素更强的抗菌性,但由于在其侧链的生产中使用了甲磺酰氯[5],会与残留的溶剂醇生成甲磺酸酯,因此需要对替比培南酯侧链中的甲磺酸酯类杂质进行检测.
目前检测甲磺酸酯的方法主要有GC-MS法[6-10]、LC-MS法[11-13]和气相色谱-微池电子捕获法[14].微池电子捕获检测器灵敏度高,但线性范围窄;质谱(MS)检测器因具有选择性强、灵敏度高、线性范围宽等优势而成为重要的检测手段[15].由于直接进样测定难以避免原料药本身对仪器的污染及离子化效率低等问题,在GC-MS方法中,使用低挥发性的衍生试剂,通过顶空萃取挥发性衍生产物可有效降低基质干扰.目前采用较多的方法是以乙腈/水(体积比80∶20)混合溶剂为衍生基质的碘化钠衍生方法[9-10],本文改进了碘化钠衍生方法,将甲磺酸酯与碘化钠在2-己酮/水(体积比3∶97)溶剂中反应得到相应的碘代烷烃产物,在优化样品的顶空萃取条件和色谱条件的基础上,实现了同时对7种甲磺酸酯类杂质的定性、定量检测,可用于活性物质中甲磺酸酯类杂质的检测.
1 实验部分
1.1 仪器与试剂
Trace ISQTM气质联用仪,配套有Triplus顶空自动进样器、顶空进样瓶(20 mL)和Xcalibur数据处理系统(EI离子源,美国Thermo Scientific公司);FiveEasy PlusTM pH计(瑞士Mettler Toledo公司);Milli-Q Reference超纯水系统(美国Millipore公司).
甲磺酸甲酯(MMS,98.0%,吴睿化学有限公司);甲磺酸正丙酯(n-PMS)和甲磺酸异丁酯(i-BMS)(>98.0%,梯希爱化成工业发展有限公司);甲磺酸仲丁酯(s-BMS)和甲磺酸正丁酯(n-BMS)(>98.0%,苏州麦凯生物医药科技有限公司);甲磺酸乙酯(EMS,98.5%)、甲磺酸异丙酯(i-PMS,97.0%)、碘化钠(99.0%)、2-己酮(99.0%)(百灵威科技有限公司);内标CHCl3(99.8%,J. T. Baker公司);硫代硫酸钠(99.0%,Sigma Aldrich公司);其他试剂均为色谱纯或分析纯.
1.2 溶液的配制
衍生试剂:称取60 g 碘化钠和30 mg硫代硫酸钠,加水溶解并稀释至50 mL,摇匀备用.
内标溶液:量取CHCl3适量,用2-己酮溶解并稀释,配制质量浓度为14.84 g/L的内标储备液,再用超纯水稀释至质量浓度为7.42 mg/L的内标溶液,摇匀备用.
甲磺酸酯类混合标准溶液:精密量取MMS、EMS、i-PMS、n-PMS、s-BMS、i-BMS、n-BMS标准品适量,用2-己酮溶解并稀释,配制质量浓度约为5 g/L的储备液,摇匀,于4 ℃冷藏备用.精密量取标准品储备液适量,用超纯水分别稀释至质量浓度约为1、1、3、2、2、2、2 mg/L的混合标准溶液,现制现用.
1.3 衍生反应
混合标准溶液的衍生:精密量取混合标准溶液200 μL置于20 mL顶空瓶中,加入超纯水320 μL、2-己酮30 μL、内标溶液50 μL和衍生试剂400 μL,立即加盖密封,涡旋混匀,于60 ℃下反应20 min.
样品溶液的衍生:精密称取原料药25 mg置于20 mL顶空瓶中,加入超纯水520 μL和2-己酮30 μL,涡旋使其溶解,再加入内标溶液50 μL和衍生试剂400 μL,立即加盖密封,涡旋混匀,于60 ℃下反应20 min.
1.4 色谱-质谱条件
色谱柱为TR-wax MS柱(30 m×0.25 mm,0.25 μm);程序升温:40 ℃保持6 min,然后以20 ℃/min升至240 ℃,保持5 min;载气为高纯氦气;流速为0.8 mL/min,恒流模式;进样口温度200 ℃;进样量100 μL;进样方式为分流进样,分流比50∶1;孵化室温度80 ℃,平衡时间为30 min.质谱条件:离子源温度250 ℃;传输线温度250 ℃;EI离子源能量70 eV;检测模式为选择离子扫描(SIM).定量监测离子:m/z142(碘甲烷),m/z156(碘乙烷),m/z170(碘代正丙/异丙烷),m/z184(碘代正丁/异丁/仲丁烷);m/z83(CHCl3).定性监测离子:m/z127(碘代烷烃),m/z118(CHCl3).
2 结果分析与讨论
2.1 衍生反应条件
2.1.1反应溶剂和用量 目前的碘化钠衍生方法均是以乙腈/水(体积比80∶20)混合溶剂为衍生基质,衍生结束后经顶空萃取,大量的强极性乙腈进入仪器中会产生很强的溶剂峰,不仅降低离子源灯丝的寿命,还容易引起色谱柱的填料流失.因此,对一些出峰位置不干扰检测物目标组分的有机相进行了考察.按1.3所述过程操作,其他条件不变,分别加入30 μL的有机相苯甲醚、氯苯、正己烷、甲基叔丁基醚、乙酸正丁酯、2-庚酮、正丁醇、2-己酮,与未加入有机相的各组分对比分析,发现2-己酮效果最好.同时对2-己酮的用量也进行了优化,确定2-己酮/水(体积比3∶97)的最佳用量为30 μL.
2.1.2衍生试剂用量 分别加入0.2、0.3、0.4、0.5 mL衍生试剂,按1.3所述过程操作,通过调整超纯水用量使顶空瓶内的液体总量保持在1 mL.结果表明,当衍生试剂加入0.4 mL时可达到最佳衍生效果,略低于文献[9-10]中0.5 mL的用量.
2.1.3衍生温度和时间 按1.3所述过程操作,分别在40、50、60、70 ℃下衍生反应20 min后进样测定,发现60 ℃下衍生效果最佳.在此基础上,考察了反应时间10、20、30、40 min对衍生反应的影响,确定最佳反应时间为20 min.
2.2 顶空萃取条件
将衍生后的样品分别在50、60、70、80 ℃下平衡30 min后顶空进样分析.结果发现,随着温度的升高,所检测到的组分含量逐渐增大,其中相对分子质量高的化合物受温度的影响程度较大,考虑到顶空瓶内压力和气密性等问题,选择在80 ℃下平衡.在此基础上,考察了在80 ℃下平衡时间对分析结果的影响,结果表明,平衡30 min时萃取效果最好,因此将顶空萃取条件设为80 ℃下平衡30 min.
2.3 混合标准溶液和衍生产物的稳定性
将新制的甲磺酸酯类混合标准溶液于室温下放置,依次取放置0、20、40、60、80 min的样品进行分析,发现在20 min内各个组分可保持稳定,超出20 min后,s-BMS最容易分解,其次是i-PMS和i-BMS.对衍生产物的稳定性进行考察,4个由混合标准溶液配制的样品衍生后于室温下放置,每隔24 h检测1个样品,发现碘代烷烃产物在3 d内都能保持稳定.
2.4 方法学验证
2.4.1线性范围、精密度和回收率 将甲磺酸酯类混合标准溶液稀释至7个质量浓度,以目标物和内标的峰面积比对样品质量浓度进行线性回归,得到线性回归方程及相关系数.通过混合标准溶液逐级稀释,由信号响应信噪比(S/N)来确定检出限(S/N=3)和定量限(S/N=10),结果如表1所示.分析物标准曲线的线性相关系数均大于0.999 0,检出限为0.21~2.64 μg/L,定量限为0.70~8.81 μg/L,对应的实际样品的检出限为8.4~106 ng/g,定量限为28~352 ng/g.取混合标准溶液(MMS、EMS、i-PMS、n-PMS、s-BMS、i-BMS、n-BMS的质量浓度分别为10、9、27、18、22、22、18 μg/L)进行日内、日间平行分析,经过精密度计算,相对标准偏差(RSD,n=3)为0.95%~10.3%,满足方法学的要求.称取替比培南酯中间体样品25 mg(含微量MMS、EMS和n-PMS杂质),分别加入混合标准溶液(MMS、EMS、i-PMS、n-PMS、s-BMS、i-BMS、n-BMS的质量浓度分别为10、9、27、18、22、22、18 μg/L)进行回收率试验,各目标物的加标回收率为88.2%~115.1%(n=3),满足方法学的要求.

表1 甲磺酸酯类化合物的线性范围、回归方程、相关系数、精密度和回收率Tab.1 The linear range, regression equation, correlation coefficient, precision and recovery of methanesulfonate compounds
2.4.2低级醇的干扰性实验 文献[9-10]中提出,空气中的甲醇会与碘化钠衍生试剂发生亲核取代反应,生成碘甲烷,进而对检测准确性造成一定的影响.而在本方法中,未见空白样品中有甲醇带来的干扰.作为补充,按照混合标准溶液的配制方法配制了甲醇和乙醇的混合溶液,于顶空瓶中的质量浓度均为127 μg/L,按同样的方法进行衍生分析,也未观察到碘甲烷或碘乙烷谱峰的出现,表明空气或样品中残留的甲醇或乙醇对该方法的检测结果没有影响.文献[9-10]中的干扰可能是由于使用了大量有机相溶剂乙腈,促进了干扰产物的生成.
2.4.3样品分析 按上述方法对某药企生产的5批替比培南酯中间体进行分析,其SIM色谱图如图1所示.由图1(a)可见,7 min内所有目标化合物均已出峰,内标CHCl3(IS)的保留时间为5.47 min,对应MMS、EMS、i-PMS、n-PMS、s-BMS、i-BMS、n-BMS的衍生物的保留时间分别为2.25、2.77、3.06、4.05、 4.91、5.17、6.43 min.图1(b)中保留时间为3.68、3.80 min的峰来源于溶剂本底.图1(c)为实际样品的SIM色谱图(图1(c)中的小图分别为实际样品中m/z142、m/z156和m/z170的萃取离子流图),均检出MMS、EMS和n-PMS,来源于生产中甲醇、乙醇、丙醇等溶剂与甲磺酰氯的副反应,而生产中未用到的异丙醇和正、异、叔丁醇没有检测到,5批样品(S1~S5)的测定结果见表2.
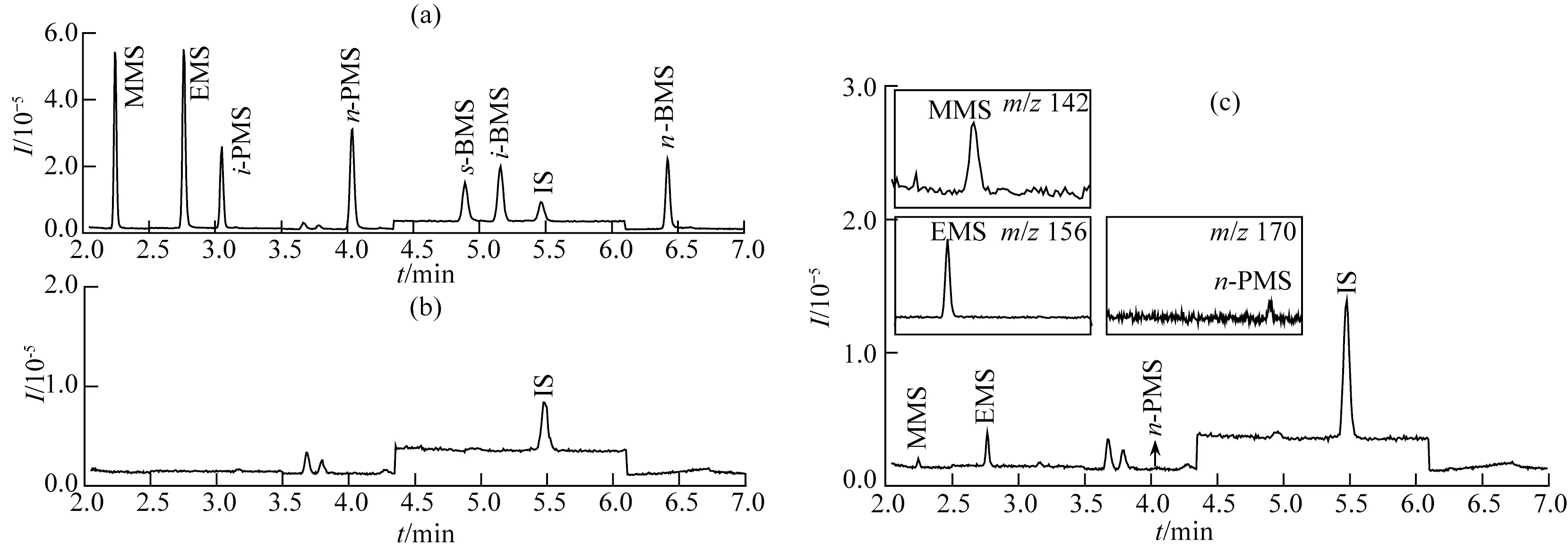
图1 混合标准品溶液(a)、空白溶液(b)和实际样品(c)的SIM色谱图Fig.1 Selected-ion monitoring chromatograms of mixed standard solution (a), blank solution (b) and the sample (c)

样品S1S2S3S4S5MMS0.1810.1990.1760.2130.188EMS0.2780.4380.4110.3740.414n-PMS0.0860.1430.0870.1400.153
3 结论
文献[8,12]中报道的色谱-质谱法对药物中甲磺酸酯类杂质同时检测的数量最多有4种,本文方法可以同时对7种甲磺酸酯类杂质进行定性、定量分析,具有更广泛的适用性.与基于碘化钠衍生的GC-MS法相比,该方法避免了有机相乙腈的大量使用,仅使用少量2-己酮就可达到分析要求.由于衍生反应溶剂中有机相用量少,避免了空气或样品中残留的甲醇或乙醇对检测结果的影响.该方法具有适用范围广、灵敏度高、基质干扰小等特点,可用于药物中间体、原料药及成品药中甲磺酸酯类杂质的检测.
