Fe,Co,Ni掺杂石墨烯表面吸附C2H4的第一性原理研究
2019-09-17宋述鹏贾娜娜龚铁夫周和荣
宋述鹏, 贾娜娜, 龚铁夫, 周和荣, 吴 润
(1. 武汉科技大学省部共建耐火材料与冶金国家重点实验室, 武汉 430081; 2. 武汉科技大学材料与冶金学院, 武汉 430081)
1 引 言
随着计算机硬件方面的不断发展,第一性原理在材料研究中的应用也越来越多,计算精度得到了很大提高,计算时间大大减少[1]. 石墨烯(Graphenc)是一种新型的碳基材料,是碳原子以sp2杂化呈蜂巢晶格排列构成的单层二维晶体,其表现出优异的电学、光学、热和机械性能. 它独特的电子特性在微电子领域和基础物理学中有广泛的应用价值. 石墨烯的问世引起了全球科学家的关注,已成为物理学界与材料科学界最热门的研究主题之一[2]. 石墨烯可以通过化学或者物理方法进行修饰或改性可以改善石墨烯的性质,拓宽石墨烯的应用领域[3]. 为了进一步提升石墨烯的性能,对石墨烯进行掺杂是一种有效的方法. 掺杂可以打开石墨烯的能带隙,掺杂原子能影响石墨烯的酸碱特性,改变电化学性能和催化性能[4]. Schedin[5]等首先发现,用石墨烯制备的传感器可以检测到单个分子在石墨烯表面的吸附和解吸附行为,这引起了科学界的极大关注,所以大量学者纷纷开始了石墨烯与气体吸附作用机制的研究. 目前研究发现石墨烯对特定气体有较强的吸附作用,对石墨烯进行掺杂和功能化可以提高其对特定气体的吸附能力和灵敏度. 有学者对石墨烯吸附甲醛进行了第一性原理计算的研究,结果发现掺Pt石墨烯对甲醛吸附能力最好[6]. 石墨烯作为新兴的功能材料,近年来在气体传感器上得到了较大的发展,而乙烯(C2H4)作为一种植物激素气体,能够被石墨烯传感器探测,然而石墨烯以及掺杂石墨烯吸附C2H4方面的理论研究较少. 因此,本文通过第一性原理计算对比和分析了本征石墨烯以及Fe、Co、Ni掺杂石墨烯表面吸附C2H4的作用过程,以期望对C2H4检测提供一种新的思路,并且可以为石墨烯材料的表面气体吸附及相关气敏元器件研究提供理论支持.
2 计算方法与结构模型
本文所有的计算都采用基于平面波的密度泛函理论DFT:(Density Functional Theory)的第一性原理下的DMol3软件包,利用密度泛函理论在广义梯度近似(Generalized Gradient Approximation, GGA)下对5×5×1石墨烯的结构进行了设计与优化,选择带极化的双数值原子基组和自旋非限制近似求解自洽场. 电子关联为PW91泛函,基组为双数值轨道基组+p轨道极化函数DNP[7],在计算过程中,电子结构的计算以体系的能量是否收敛为判据,精度优于10-5a.u.,结构优化以梯度和位移和能量是否收敛为判据,梯度和位移的收敛精度优于10-3a.u.,能量收敛精度优于10-5a.u.. 为了验证所用方法的有效性,我们首先计算了本征石墨烯的C-C键长(1.42Å)与文献中(1.42 Å)[8]符合的很好,说明本文所用方法对该体系是合适的.
3 结果与讨论
3.1 几何结构
为了对比掺入Fe、Co、Ni前后石墨烯的一些特性的变化我们分别构建了掺入前后石墨烯的几何构型,并进行了优化,得到的几何结构如图1所示,本文中所有白色原子代表H原子,灰色代表C原子,红色代表Fe原子,蓝色代表Co原子,黄色代表Ni原子.
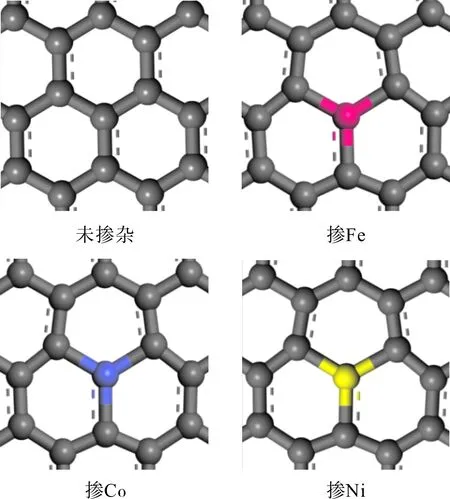
图1 Fe、Co、Ni掺杂前后石墨烯的几何构型Fig. 1 The geometrical structures of graphene (before and after Fe, Co and Ni doped)
Table 1 The structure changes ofgraphene (before and after Fe, Co and Ni doped)

未掺杂掺杂Fe掺杂Co掺杂NidC-M(Å)1.421.661.661.42S(Å2)5.245.775.755.28
(注:dC-M:C-M键长,M=Fe、Co、Ni;S:掺杂前后六边形的面积)
首先考虑原子掺杂对石墨烯的影响,杂质原子 Fe、Co、Ni为第四周期元素,原子半径比 C 原子大,不易与 C 原子形成杂化,杂质周围C原子也发生移动[9~10]. 由表1可以看出掺杂Fe、Co后变形较大,六边形的面积分别由原来的5.24 Å2变为了5.77 Å2,5.75 Å2分别变化了0.53 Å2,0.51 Å2,而掺杂Ni六边形面积变化较小变为了5.28 Å2变化了约0.04 Å2,Fe、Co、Ni原子到最近邻碳原子的距离分别为dFe-C=1.66 Å,dCo-C=1.66 Å,dNi-C=1.42 Å,其中Fe-C和Co-C 键长最长,Ni-C键和C-C 键相比无变化,由此可见掺杂Fe、Co、Ni后石墨烯的比表面积增大,便可提供更多的活性位点[11],使得电子迁移率增大,这种结构的变化对石墨烯的活性会产生一定影响.
3.2 乙烯吸附的影响
3.2.1本征石墨烯
为了寻找石墨烯体系最佳的吸附位置,对3种不同的吸附位置都分别进行了考虑,如乙烯在本征石墨烯的顶位(T)、桥位(B)和心位(H)的吸附结构模型和DOS图分别如图2和3所示:
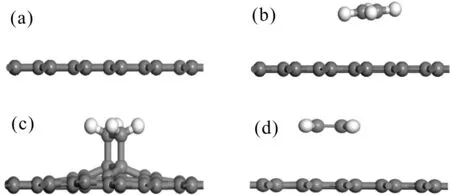
图2 本征石墨烯吸附乙烯的结构模型((a)未吸附,(b)T位吸附,(c)B位吸附,(d)H位吸附)Fig. 2 Structural models of C2H4 adsorption on intrinsic graphene ((a) No adsorption, (b) T site position, (c) B site position, (d) H site position)
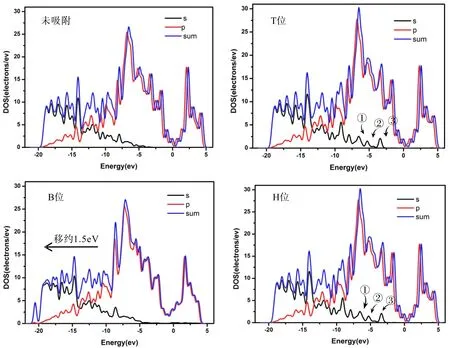
图3 本征石墨烯吸附乙烯前后的DOSs((a)未吸附,(b)T位吸附,(c)B位吸附,(d)H位吸附)Fig. 3 The DOSs of C2H4 adsorption on intrinsic graphene((a) No adsorption ,(b) T site position, (c) B site position, (d) H site position)
由图2可以看出乙烯在本征石墨烯T位和H位的吸附对结构并没有影响,而在B位的吸附会一定程度破坏本征石墨烯基底,使得与乙烯吸附的C原子高于本征石墨烯基底平面,这可能使乙烯在本征石墨烯的B吸附时乙烯与本征石墨烯的C=C双键断裂发生缩聚反应,由此可知乙烯在本征石墨烯B位的吸附为化学吸附,对基底的影响作用较大. 从图3可以看出本征石墨烯在费米能级处,态密度接近于零,为半金属特性[12,13],吸附乙烯后在B位吸附会使得态密度整体向导带移动约1.5 eV而在T位和H位的吸附使得S轨道在-2.5~-7.5 eV之间出现三个小尖峰(见图3中①、②、③处),同时使总的态密度有所升高.
3.2.2掺杂石墨烯
图4(a)-(c)分别是掺Fe、Co和Ni石墨烯未吸附和吸附乙烯后的结构图. 由图4可知吸附乙烯后都会影响掺杂石墨烯基底,使得Fe、Co、Ni原子及与掺杂原子周围相连的C原子位置都高于掺杂石墨烯基底平面,由此可以得出掺杂石墨烯的吸附为化学吸附. 图4(a)优化后乙烯在T、B、H位的初始吸附位最终都会趋向于B位的吸附,说明乙烯在掺Fe石墨烯的最佳吸附位置为B位吸附,而图4(b)-(c)优化后乙烯都趋向于H位的吸附,说明乙烯在掺Co、Ni石墨烯的最佳吸附位为H位吸附.

图4 掺杂石墨烯吸附乙烯前后的结构模型((a)掺Fe石墨烯,(b) 掺Co石墨烯,(c) 掺Ni石墨烯)Fig. 4 Structural models of C2H4 adsorption on doped graphenes((a) Fe-doped graphene,(b) Co-doped graphene, (c) Ni-doped graphene)
为了进一步理解石墨烯掺杂过渡族元素时的变化,我们对比分析了本征石墨烯以及掺杂石墨烯的分波态密度(PDOS). 比较图3和图5可以看出由于Fe、Co、Ni的加入使得DOS图均出现d轨道,从图5可以看出掺杂石墨烯与本征石墨烯相比DOS图都会整体向导带方向偏移约0.2~1.0 eV,而且最高峰也有所降低,在图5中-20~-15 eV能量区间内DOS分布较为平均、没有出现局域尖峰,说明对应的是类sp带,表明电子的非局域化性质很强. 由图5可以看出在掺杂石墨烯的T、B、H位吸附乙烯后会使得其态密度-5~-6 eV处的S轨道出现一个小尖峰(图5黑色箭头所指). 图3和图5均显示掺杂后费米能级处的态密度都会增高,这主要来自于所掺杂金属d轨道的贡献,使得掺杂石墨烯表现出金属特性. 从图5中吸附前后对比可以看出掺杂Fe后的费米能附近的态密度增高的最多,Co次之,Ni较少,因此掺杂Fe后石墨烯的金属特性最显著. 费米能级附近的态密度对体系导电性的影响比较大,比较图3和图5可以看出掺杂后将使费米能级附近的态密度积分显著提高,费米能级附近的积分直接影响到电子的态密度数,从而很大程度上影响掺杂石墨烯的电导性质,这在传感器的研究方面具有重要作用[14].
3.2.3吸附能
吸附能的大小表征了材料中原子间所成键的强弱,吸附能越高表示该体系结构越稳定,通过比较不同结构吸附能的大小就能够判定一定热力学条件下结构的相对稳定性[15]. 因此,为了考察吸附C2H4后的稳定性,我们计算了体系的吸附能,通过比较不同结构的吸附能高低来讨论体系的结构稳定性.
C2H4与石墨烯(G)之间的吸附能定义为:
Ead=E(G-M)+EC2H4-E(C2H4@G-M)
(1)
其中,E(C2H4@G-M)表示吸附C2H4分子后体系的总能量,E(G-M)表示掺杂石墨烯的总能量,EC2H4是乙烯的总能量.
由表2可以看出乙烯在本征石墨烯B位上的吸附能高于在本征石墨烯T和H位上的吸附,这在其吸附模型和态密度图中都有体现,说明乙烯在本征石墨烯B位的吸附较为稳定;而乙烯在本征石墨烯上的吸附能明显低于掺杂Fe和Ni时的吸附能,由此可见,乙烯在掺杂Fe和Ni石墨烯基底上的吸附相对较为稳定,且掺杂Ni时体系最稳定,而掺杂Co最不稳定,由表2还可以看出乙烯在本征石墨烯上吸附时,在T位和B位上的吸附能高于在掺Co石墨烯上T位和B位的吸附能,在掺杂Fe和Ni的石墨烯上吸附乙烯时在T位和B位最稳定,而掺杂Co石墨烯在H位的吸附最稳定.
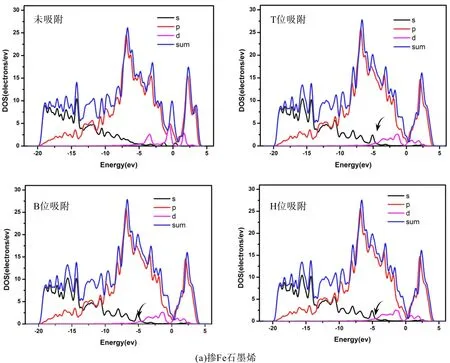
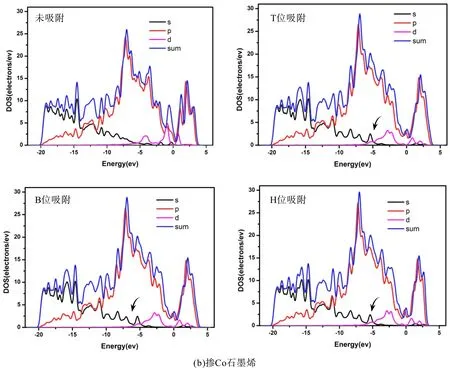
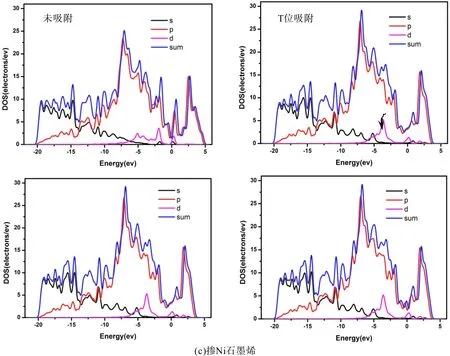
图5 掺杂石墨烯吸附乙烯前后的PDOS图((a) 掺Fe石墨烯,(b) 掺Co石墨烯,(c) 掺Ni石墨烯)Fig.5 The P DOS of C2H4 adsorption on intrinsic graphenes((a) Fe-doped graphene,(b) Co-doped graphene, (c) Ni-doped graphene)
表2 掺杂前后石墨烯的吸附能(eV)
Table 2 Adsorption energy of graphene before and after doping
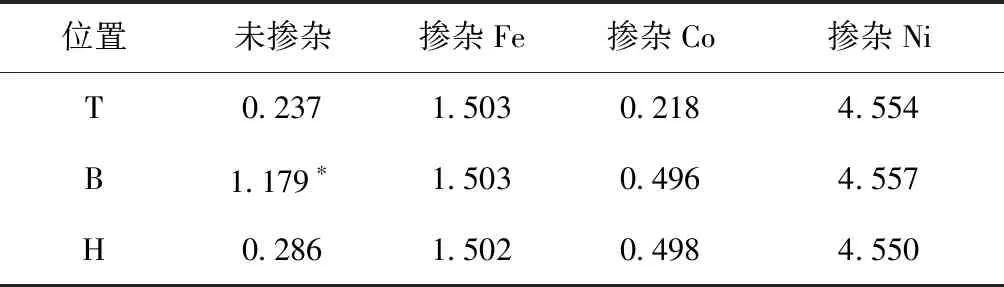
位置未掺杂掺杂Fe掺杂Co掺杂NiT0.2371.5030.2184.554B1.179∗1.5030.4964.557H0.2861.5020.4984.550
(*本征石墨烯B位吸附能为绝对值)
4 结 论
采用基于密度泛函理论的第一性原理计算方法, 研究了本征石墨烯及掺杂石墨烯表面对C2H4分子的吸附行为,研究结果如下:
(1)掺杂Fe、Co、Ni后石墨烯的比表面积增大,便可提供更多的活性位点增多,使的电子迁移率增大;
(2)乙烯在本征石墨烯B位的吸附和掺杂石墨烯的吸附为化学吸附,在本征石墨烯T和H位的吸附为物理吸附;
(3)乙烯在掺Fe、Ni石墨烯的最佳吸附位为T位和B位,在掺Co石墨烯的最佳吸附位为H位;掺杂Fe、Ni后体系的吸附能力显著提高,且掺杂Ni时体系的吸附能力最好.
