介导缺氧诱导因子-1α表达的相关信号通路在类风湿性关节炎中的研究进展
2019-09-13孙明慧卜妍红戴学静
孙明慧,吴 虹,卜妍红,张 衡,戴学静,王 言,占 翔
(安徽中医药大学药学院,新安医学教育部重点实验室,中药复方安徽省重点实验室,安徽 合肥 230012)
类风湿性关节炎(rheumatoid arthritis,RA)是一种常见的、以慢性滑膜炎症为临床表征的自身免疫性疾病,其病理特征是滑膜组织异常炎性增生、血管新生、血管翳形成、关节软骨和骨的不可逆性破坏,甚至引发畸形病变以及严重的并发症[1]。迄今为止,RA病因尚未明确,但已有研究表明,缺氧微环境作为RA滑膜炎症组织的重要特征之一,参与调节滑膜炎症反应、血管新生和软骨破坏等关键病理生理过程[2]。
缺氧能促进炎性因子的释放,加重炎症反应;反之,炎症也能加重组织缺氧。缺氧诱导因子1α (hypoxia-inducible factor-1α,HIF-1α)是调节细胞对缺氧反应的核转录因子,在缺氧微环境中,HIF-1α水平明显上升,并调节胞内许多基因的转录与表达。血管内皮生长因子(vascular endothelial growth factor,VEGF)是最具特征性的HIF-1α下游靶基因之一,缺氧导致HIF-1α积累,并触发HIF-1α和VEGF途径的正反馈调节,促使血管生成以改善机体低氧环境,而降低HIF-1α水平可通过VEGF依赖机制抑制血管生成[3-4]。HIF-1α还参与诱导基质金属蛋白酶(matrix metalloproteinases,MMPs)的分泌,调控RA成纤维滑膜细胞(fibroblast-like synoviocytes,FLSs)的迁移和侵袭机制,阻断HIF-1α表达可抑制缺氧诱导的RA-FLSs迁移和侵袭,改善滑膜增生及软骨和骨破坏[4-5]。
随着细胞信号转导通路地深入研究,越来越多RA病理调节机制被发现。目前,除氧感受器通路脯氨酸羟化酶(prolyl hydroxylases,PHDs)-HIFs-pVHL外,已发现调控HIF-1α表达的信号通路还包括PI3K/Akt、Ras-Raf-MEK-ERK、JAK/STAT、NF-κB等,这些细胞信号转导通路中的效应蛋白有望成为治疗RA潜在的作用靶点。鉴于HIF-1α对RA病情的推动作用,笔者对调控HIF-1α表达的信号通路进行综述,以期从改善病情角度为RA治疗及抗RA新药研发提供思路。
1 PHDs/HIF-1α/pVHL通路
PHDs属于氧依赖性羟化酶,包括3个亚型:PHD1~3,其活性随着细胞内氧含量的变化而变化,对HIF-1α的稳定性调控起关键作用。HIF-1α含有1个氧依赖性的降解结构域(oxygen dependent degradation domain,ODDD),在常氧条件下,该结构域中的脯氨酸残基Pro402和Pro564极易被氧敏感的PHDs羟基化,并与肿瘤抑制蛋白pVHL结合,聚集多种泛素蛋白(ubiquitin,Ub),组成泛素蛋白酶复合体,HIF-1α被蛋白酶体快速降解。除PHDs之外,天冬酰胺羟化酶作为HIF-1抑制因子(factor-inhibiting HIF-1,FIH-1),可将HIF-1α亚基C-末端反式激活结构域(transactivation domain,TAD)中的天冬酰胺残基Asn803羟基化,干扰其与辅因子p300/CBP的结合。在低氧条件下,PHDs和FIH-1活性迅速降低,导致HIF-1α在细胞质中降解受阻,并大量聚集、积累并活化,继而转移至细胞核内,与结构亚基HIF-1β及辅因子p300/CBP发生聚合,形成HIF-1α复合物。该复合物与特定的DNA序列结合,进而与靶基因上的缺氧反应元件(hypoxia-responsive elements,HREs)结合,调控下游靶基因的激活和转录。缺氧引起的氧感受通路的异常活化,在RA病程中发挥重要作用(Fig 1)[6]。
在RA关节缺氧环境中,氧依赖性羟化酶在FLSs中短暂沉默,进而上调HIF-1α的水平,诱导许多促血管生成因子和炎症介质的表达。Muz等[7]发现,沉默PHD1或FIH-1后,HIF-1α水平无影响,而沉默PHD2可增强HIF-1α的稳定性,诱导下游多种靶基因的表达(包括促血管生成基因等),表明PHD2是调节RA-FLSs中PHDs/HIF-1α/pVHL通路的关键参与者。对比缺氧条件下HIF-1α与促血管生成基因的表达水平发现,PHD2沉默引起的变化与缺氧条件非常相似,意味着PHD2的缺失可以模拟缺氧环境,但在正常滑膜细胞中,沉默PHD2不影响HIF-1α的稳定性,其具体调控机制仍需进一步研究。结缔组织生长因子(connective tissue growth factor,CTGF)属于富含半胱氨酸生长因子家族的一员,其作为一种炎性介质在RA中高表达,CTGF的水平与VEGF表达呈正相关。进一步研究发现,CTGF可通过上调microRNA-210的水平,抑制甘油-3-磷酸脱氢酶1样蛋白(glycerol-3-phosphate dehydrogenase 1-like,GPD1L)表达,导致PHD活性降低,HIF-1α积累,进而上调VEGF mRNA水平及VEGF的表达,促进血管生成[8]。
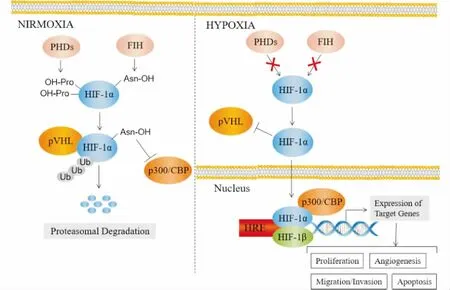
Fig 1 Mechanism of PHDs/HIF-1α/pVHL signaling
Under normoxic conditions, PHDs and FIH hydroxylate different sites of HIF-1α, respectively, producing pVHL binding sites, blocking the combination with cofactor p300/CBP, and resulting in HIF-1α degradation. Under hypoxic conditions, the activity of PHDs and FIH decreased, HIF-1α accumulates and migrates into the nucleus to form a complex with HIF-1β and p300/CBP. The complex combines with HRE to induce target gene transcription, and then regulates cell proliferation, migration/invasion, apoptosis and angiogenesis.
2 PI3K/Akt信号转导通路
磷脂酰肌醇-3激酶(phosphoinositide 3-kinase,PI3K),属于胞内磷脂酰肌醇激酶,是细胞中重要的信号调节蛋白,其同时具有丝氨酸/苏氨酸(Ser/Thr)激酶和磷脂酰肌醇激酶的活性。PI3K分为结构与功能各异的3型,其中I型PI3K研究最广泛,由催化亚基p110和调节亚基p85构成。Akt作为PI3K下游主要的效应分子之一,也是一类特异性的Ser/Thr蛋白激酶。PI3K/Akt信号转导通路在滑膜细胞增殖和凋亡失衡中发挥关键作用,其异常活化引起滑膜过度增生并向软骨和骨组织浸润生长,诱导血管新生及血管翳形成,导致关节畸形和骨破坏[9-10]。
许多信号蛋白分子和信号传导复合物都能启始PI3K的激活过程,诱发磷脂酰肌醇二磷酸(phosphatidylinositol diphosphate,PIP2)磷酸化,产生第二信使三磷酸磷脂酰肌醇(phosphatidylinositol triphosphate,PIP3),PIP3与存在PH结构域的蛋白激酶Akt相互作用,导致Akt构型改变,形成Ser473和Thr308的磷酸化位点,并移位至细胞膜。同样存在PH结构域的3-磷酸肌醇依赖性蛋白激酶-1(3-phosphoinositide dependent protein kinase-1,PDK1)与哺乳动物雷帕霉素靶复合体2(mammalian target of rapamycin complex 2,mTORC2)分别将Akt上的Ser473和Thr308位点磷酸化,致使Akt被激活(Fig 2)。
哺乳动物雷帕霉素靶向蛋白(mammalian target of rapamycin,mTOR)是PI3K/Akt信号通路的下游效应分子之一。张晓军等[11]采用弗氏完全佐剂建立佐剂性关节炎(adjuvant arthritis,AA)大鼠模型,采用ELISA法检测大鼠血清中HIF-1α、VEGF的表达,Western blot检测滑膜组织PI3K、Akt1、p-Akt1、mTOR蛋白表达情况发现,与正常组比较,模型组血清VEGF、HIF-1α和滑膜组织PI3K、Akt1、p-Akt1、mTOR表达明显升高,表明活化的PI3K/Akt/mTOR信号转导途径可能参与诱导滑膜血管新生。Zhang等[12]探讨低氧条件下RA-FLSs活性、细胞凋亡和侵袭性的影响发现,与常氧细胞相比,缺氧RA-FLSs侵袭性明显增强,且其侵袭能力与HIF-1α表达水平呈正相关。曲古抑菌素A可明显抑制缺氧诱导RA-FLSs的活力,诱导细胞凋亡,阻断RA-FLSs侵袭,并降低MMP-2、MMP-9的表达。进一步对其作用机制进行探究发现,这些作用通过下调PI3K活性,进而抑制Akt活化实现,而活性Akt的过表达可逆转其对RA-FLSs侵袭性及MMP-2、MMP-9下调的抑制作用,表明缺氧诱导的RA-FLSs中的促凋亡与抗侵袭活性与PI3K/Akt信号转导的激活有关。
3 Ras/Raf/MEK/ERK信号转导通路
作为MAPKs的一个亚族,胞外信号调节激酶(extracellular signal-regulated kinase,ERK)包括5个亚型,即ERK1~ERK5,其中对ERK1/2研究最为广泛。在炎性环境中,许多细胞因子参与启始Ras/Raf/MEK/ERK信号转导通路,通过一系列复杂过程被细胞表面受体和胞内传感机制活化,参与多种生物学反应。ERK的异常活化除介导RA中细胞增殖和炎症反应之外,还参与调控血管生成、软骨及骨破坏等过程,导致RA病情不断恶化[13]。
多数信号因子对ERK1/2的活化都始于对Ras的激活,Ras是信号传递的“分子开关”,其被激活后作用于Raf的氨基末端,使Raf从胞质移位至细胞膜上,通过丝/苏氨酸残基磷酸化而活化。Raf激活后,其C端催化区可与MEK结合,并使两个丝氨酸磷酸化,进而激活MEK,随后通过酪氨酸和苏氨酸双特异性磷酸化激活ERK,形成Ras/Raf/MEK/ERK途径(Fig 2)。
研究证实,炎症因子、生长因子和某些细胞因子受体均可激活ERK信号转导通路,B细胞活化因子(B-cell activating factor,BAFF)属于肿瘤坏死因子超家族成员之一,在B细胞的成熟和维持中起作用,且BAFF与自身免疫疾病密切相关。Lee等[14]探究了TNF-α诱导的BAFF表达对RA-FLSs和人类风湿性关节炎成纤维样滑膜细胞MH7A存活的影响,通过使用BAFF siRNA转染细胞,证实BAFF表达与细胞存活率呈正相关。常氧条件下,TNF-α诱导的MH7A细胞BAFF、VEGF和HIF-1α的转录及表达可被HIF-1α siRNA阻断;缺氧条件下或过表达HIF-1α时,HIF-1α siRNA阻断作用被逆转。进一步研究发现,ERK抑制剂PD98059抑制TNF-α诱导的BAFF表达,而HIF-1α的过表达可使PD98059抑制作用被逆转,表明TNF-α通过ERK依赖的HIF-1α表达来调控FLSs的存活。ERK的激活还参与调节血管新生,来源于三七叶片的三七皂苷Ft1可通过激活ERK信号通路,诱导人脐静脉内皮细胞(human umbilical vein endothelial cells,HUVECs)增殖、迁移,诱导管形成。对其具体机制探究发现,Ft1刺激Raf/MEK/ERK途径磷酸化,促进HIF-1α从胞质向胞核的转运及HIF-1α与VEGF启动子的结合,上调VEGF mRNA的转录水平及VEGF的分泌。Ft1也参与调控PI3K/Akt/mTOR途径磷酸化。进一步研究发现,mTOR及其效应器核糖体40S小亚基S6蛋白激酶(P70S6K)是PI3K/Akt和Raf/MEK/ERK的下游靶点,沉默mTOR致使Ft1的促血管生成作用被抑制,表明PI3K/Akt和Raf/MEK/ERK通路均通过下游mTOR/P70S6K途径,调控HIF-1α的活化及VEGF的转录与表达[15]。
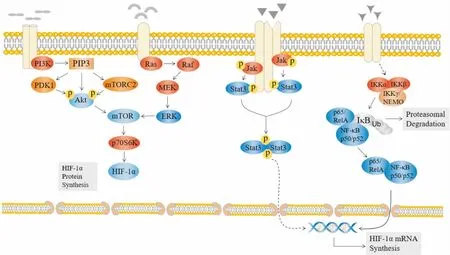
Fig 2 Mechanism of signaling pathways regulating HIF-1α mRNA and protein expression
Inflammatory factors activate PI3K and induce PIP3production. Under the effect of PKD1 and mTORC2, Akt is phosphorylated and activated. After inflammatory factors stimulated Ras, Raf/MEK phosphorylate and finally activate ERK. Both activated Akt and ERK can phosphorylate mTOR and activate its effector protein p70S6K, increasing the translation level of HIF-1α.
Inflammatory factors bind to its receptors on the membrane and then activate the JAK/STAT signaling pathway. The activated STAT proteins are translocated into the nucleus to bind to the target genes in the form of a dimer, thus affecting the synthesis of HIF-1α mRNA. Inflammatory factors activate IKK which degrades IκB, and resulting in activated NF-κB releases. Activated NF-κB transports into the nucleus and binds to target gene, finally induces the synthesis of HIF-1α mRNA.
4 JAK/STAT信号转导通路
JAK属于典型的非跨膜酪氨酸激酶,JAK家族由4个成员组成,分别是JAK1-JAK3以及Tyk2,其可使与其相结合的细胞因子受体磷酸化,它还能够使含有SH2结构域的蛋白分子发生磷酸化反应。STAT被称为“信号转导子和转录激活子”,目前为止已发现STAT家族的7个成员,STAT蛋白包含6个功能区段,其中SH2结构域是最重要的功能区段,可与细胞因子受体酪氨酸磷酸化特异性结合。目前,大量的研究表明JAK/STAT信号通路功能失调可诱导RA-FLSs增殖,导致滑膜增生;调节免疫反应,造成滑膜炎症;调控靶基因MMPs的分泌,加重软骨及骨破坏[16]。
JAK-STAT信号转导通路通常由诸多细胞因子激活,介导细胞多种生物学功能,在免疫调节和炎症反应过程中发挥关键作用。首先,信号分子作用于相应的受体后,与受体偶合的JAK激酶发生酪氨酸磷酸化作用并被活化。然后,活化的JAK促使受体发生磷酸化修饰,产生酪氨酸磷酸化位点;同时,含有SH2结构域的STAT蛋白被募集至该位点,经JAK催化磷酸化修饰后,磷酸化的STAT蛋白以二聚体形式移位至细胞核内,与靶基因结合并诱导其转录与翻译(Fig 2)。
为探讨低氧对RA中STAT3诱导的促炎途径的影响,甄晓洲等[17]通过建立低氧诱导的THP-1细胞的炎症模型发现,低氧可促进炎症因子IL-18、TNF-α的分泌,并提高细胞内p-JAK2、p-STAT3蛋白的表达。JAK2抑制剂AG490能明显抑制低氧诱导的p-JAK2、p-STAT3的蛋白表达,降低促炎因子IL-18、TNF-α的分泌。即低氧可能通过激活JAK2/STAT3信号通路,诱导细胞炎症因子的表达增加,加重炎症反应。Gao等[18]研究发现,缺氧诱导滑膜组织HIF-1α与p-STAT1/3的表达,并促进p-STAT3核易位,该效应可被STAT3 siRNA和JAK2抑制剂WP1066阻断;且缺氧诱导的细胞侵袭、迁移和细胞因子产生受到STAT3 siRNA和WP1066的抑制。HIF-1α siRNA可降低缺氧诱导的p-STAT3水平,STAT3 siRNA也能抑制缺氧诱导的HIF-1α表达水平,表明在RA促炎机制的调节中,HIF-1α和STAT3信号之间存在功能联系;在体外RA滑膜移植培养中,JAK2抑制剂明显降低IL-6、IL-8和MMP3的自分泌,并诱导IL-10表达,进一步证实STAT阻断在RA治疗中的作用。目前,JAK/STAT信号途径对RA中HIF-1α表达的研究尚不多见,但在许多癌症发病机制中已进行了深入探讨,JAK/STAT信号途径的激活是通过上调HIF-1α mRNA及其蛋白的表达,抑制癌细胞凋亡、促进肿瘤血管新生以及癌细胞侵袭和转移等过程。所以我们有理由推断,在RA中,JAK/STAT信号途径亦能调控HIF-1α mRNA的表达,该机制具体途径有待进一步研究。
5 NF-κB信号转导通路
NF-κB是哺乳动物中一类关键的转录激活因子,NF-κB家族由NF-κB1(p50)、NF-κB2(p52)、RelA(p65)、RelB和cRel组成,其N端均存在Rel同源区,可与DNA结合,并特异性识别DNA碱基序列;RelA(p65)、c-Rel和RelB三者C端存在反式激活结构域,能独立诱导基因表达。NF-κB常以p65/p50二聚体形式存在。NF-κB的活化可诱导炎性介质持续作用,在细胞分化、凋亡、黏附、炎症及免疫应答等方面影响RA的发生、发展[19]。
静息状态时,抑制蛋白IκB可通过其C端特定的锚蛋白重复序列结合,并封闭NF-κB蛋白的核定位区域(nuclear-localization sequence,NLS),抑制NF-κB的活性。在经典途径中,NF-κB的活性受IκB激酶(IκB kinase, IKK)的控制,该激酶介导IκB磷酸化并降解,诱导NF-κB的核转位(Fig 2)。缺氧对RA-FLSs中NF-κB活化的影响机制尚不清楚,但在其他细胞的研究中发现,常氧条件下,IKK可被PHDs羟基化失活,抑制NF-κB的活化;在缺氧条件下,PHDs对IKK的羟化活性降低,导致IκB磷酸化并降解,进而缓解NF-κB的抑制作用[20]。
炎症可激活NF-κB信号转导途径,进而上调多种促炎因子释放,导致慢性、持续性的炎症反应,进而加重滑膜炎症,促进软骨和骨破坏以及血管翳生成。Trebec-Reynolds等[21]发现,经NF-κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)处理后,巨型破骨细胞中HIF-1α mRNA及蛋白表达增加,VEGF-A表达水平升高。二甲基双酚A作为有效的HIF-1α抑制剂,可明显降低巨型破骨细胞中的VEGF-A蛋白水平;NF-κB抑制剂胶酶毒素明显抑制RANKL诱导的HIF-1α mRNA及蛋白表达,表明RANKL通过激活NF-κB途径,提高HIF-1α mRNA及蛋白水平,高水平的HIF-1α促进VEGF表达,进而促进血管新生和骨重建循环,加重RA病情。高迁移率族蛋白1(high mobility group protein 1,HMGB1)是一种由滑膜组织中多种免疫细胞和滑膜细胞共同释放的促炎介质,Park等[22]发现,HMGB1可增强RA患者关节滑膜细胞中HIF-1α mRNA的转录水平和活性,并刺激VEGF的表达;用抗HMGB1抗体干预其作用可抑制血管新生,并降低HIF-1α的表达,降低HIF-1α水平可抑制HMGB1诱导的VEGF表达。进一步研究发现,HMGB1受体TLR4参与HMGB1诱导的HIF-1α表达,且与NF-κB的转录活性有关,抗TLR4抗体明显抑制HMGB1诱导的NF-κB p65的活性,抑制NF-κB激活可阻断HMGB1依赖的HIF-1α mRNA的表达及其活性的上调,提示NF-κB调控参与了HIF-1α基因的转录与表达。
6 其他信号转导通路
Notch信号通路由Notch受体、Notch配体及细胞内效应器分子CSL(CBF-1, Suppressor of hairless, Lag)蛋白三部分组成。Notch信号的产生通常来源于相邻细胞Notch配体与受体的相互作用,继而释放出有活性的Notch蛋白NICD(Notch intracellular domain, 又称ICN),NICD易位至细胞核内与CSL蛋白相结合,并招募核转录激活蛋白家族MAML(mastermind-like),继而形成三元络合转录复合物,其作用于Notch信号通路的相应靶点基因,发挥多种生物学作用。Gao等[23]研究发现,Notch配体DLL4、Jagged-1与NICD在RA滑膜组织中表达增加,且滑膜组织氧含量与患者滑膜组织中NICD的表达呈负相关;Notch抑制剂DAPT可以抑制HIF-1α的表达及活化,进而抑制血管新生。缺氧条件下,Notch-1还与STAT3信号之间存在功能联系,缺氧诱导的Notch-1IC、DLL4和靶基因HRT-1、HRT-2的表达均可被JAK2抑制剂WP1066抑制,即通过阻断STAT信号传导,抑制Notch信号途径的激活,进而调节RA促炎机制,达到治疗RA的目的[18]。
1-磷酸鞘氨醇(sphingosine-1-phosphate,S1P)是一种生物活性脂质,由鞘氨醇(sphingosin,Sph)经鞘氨醇激酶(sphingosine kinases,SphKs)磷酸化产生,可与细胞膜表面G蛋白偶联受体(G protein-coupled receptors,GPCRs)家族成员S1P受体(S1P receptors,S1PRs)结合,S1PRs通过调节相关炎症信号通路,影响新生血管的形成[24]。Sassoli等[25]对骨髓间充质基质细胞研究发现,S1P/S1PR1介导缺氧状态下MMP-2与HIF-1α的表达,S1PR1拮抗剂降低HIF-1α表达及其核定位,表明S1PR1介导的信号传导可能维持HIF-1α在缺氧条件下的表达及活性。
7 展望
通过对介导HIF-1α表达的相关信号通路的调研发现,HIF-1α水平及其介导的促炎、促血管新生等作用是一个受多机制、多信号通路调控的过程,且各通路之间相互交错,关联错综复杂。通过调节介导HIF-1α表达的多种信号途径,可为RA治疗提供新的方向。目前,已研究发现多种信号通路抑制剂如托法替尼(JAK抑制剂)、替西罗莫司(mTOR抑制剂)、贝伐单抗(抗VEGF抗体)等,参与抑制细胞增殖和存活,抑制炎症反应、血管生成、炎性细胞浸润和关节损伤等过程[20]。因此,从信号通路途径深入研究RA的发病机制,发掘信号通路中的关键环节和关键因子,并筛选其特异性阻滞剂阻断信号传递,以期为RA治疗和抗RA新药的研发开拓新思路。
