超高效液相色谱串联质谱法测定植物源调味料中氯虫苯甲酰胺残留
2019-09-05钱兵赵婧何燕韩丙军彭黎旭王文斌
钱兵 赵婧 何燕 韩丙军 彭黎旭 王文斌


摘 要 本研究建立了超高效液相色谱串联质谱(UPLC-MS/MS)测定植物源调味料中氯虫苯甲酰胺残留量的分析方法。调味料样品经改进后的QuEChERS方法前处理,选择C18超高效液相色谱柱进行分离,采用电喷雾离子源正离子模式、多反应监测方式进行采集,外标法定量。结果表明,方法在0.005~0.200 ?g/mL范围内线性相关系数均优于0.999,加标回收率在70%~108%之间,相对标准偏差在1.9%~10%之间。方法检出限为0.002 mg/kg,定量限为0.005 mg/kg。该方法操作简便快速、灵敏、准确性高,适用于植物源调味料中氯虫苯甲酰胺的残留量检测。
关键词 超高效液相色谱-串联质谱法;植物源调味料;氯虫苯甲酰胺
中图分类号 X839.2 文献标识码 A
Abstract A simple and efficient method based on ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) detection was developed to analyze the residue of chlorantraniliprole in plant-derived condiment. The condiment samples were pretreated with a modified QuEChERS method, and separated on an UPLC C18 column. The analyses of chlorantraniliprole were operated with electrospray ionization mass spectrometry under positive mode using a multiple reaction monitoring mode, and then quantitatived by an external standard method. The results showed that the correlation coefficients of the calibration curves were over 0.999 in linear range of 0.005–0.200 ?g/mL. The average recoveries at three spiked concentration levels varied from 70% to 108% with relative standard deviations (RSDs) of 1.9%–10%. The limits of detection (LOD) and quantitation (LOQ) was 0.002 mg/kg and 0.005 mg/kg, respectively. In conclusion, this simple and efficient method meet the method validation requirements for the determination of chlorantraniliprole residues in plant-derived condiment.
Keywords UPLC-MS/MS; plant-derived condiment; chlorantraniliprole
DOI 10.3969/j.issn.1000-2561.2019.07.030
氯蟲苯甲酰胺(chlorantraniliprole)是一种高效、低毒、广谱的新型杀虫剂[1],主要作用方式为胃毒作用和触杀作用,结构式见图1。其作用机理为激活兰尼碱受体,释放平滑肌和横纹肌细胞内储存的钙,从而引起肌肉调节衰弱、麻痹,最终导致害虫死亡[2],被广泛用于鳞翅目害虫[3]。近年来被广泛使用于蔬菜、水果和粮食作物上的虫害防治[4-5]。
图1 氯虫苯甲酰胺化学结构式
Fig. 1 Structure formula of chlorantraniliprole
随着氯虫苯甲酰胺在不同农作物生产中广泛使用,其在农产品及食品中可能引起的残留风险受到人们的广泛关注。我国《食品安全国家标准 食品中农药最大残留限量》(GB 2763-2016)规定了氯虫苯甲酰胺在谷物、油料和油脂、蔬菜、水果、坚果、糖料及调味料等不同农产品及食品中的残留限量。但是,由于我国尚未制定氯虫苯甲酰胺在农产品中残留检测方法标准,目前国家标准GB 2763-2016规定的氯虫苯甲酰胺残留限量为临时限量。因此,迫切需要建立氯虫苯甲酰胺在不同农产品及食品中的残留检测方法,为氯虫苯甲酰胺的监管提供技术支持。
目前,氯虫苯甲酰胺的检测分析方法主要使用高效液相色谱法[6]和液质联用法[7],主要涉及的农产品及食品包括谷物和油料作物[8-9]、蔬菜[10-12]、甘蔗等糖料作物[13-14]、水果[15]及动物源性食品[16]等,但是尚未见调味料等农产品中氯虫苯甲酰胺残留检测方法的报道。在已报道文献中,基于QuEChERS(Quick、Easy、Cheap、Effective、Rugged、Safe)前处理净化方法具有快速、便捷等优点,被广泛用于各类农产品及食品中氯虫苯甲酰胺的提取和净化[9-10]。但是,由于植物源调味料的特殊气味通常由硫化物、酚类、醇类化合物等构成[17-19],其基质比较复杂,使用常规QuEChERS方法净化处理后,在使用高效液相色谱-串联质谱(UPLC-MS/MS)进行分析时容易产生严重的干扰,引起回收率偏低。因此,本文利用改进的QuEChERS方法对植物源调味料进行净化处理,建立高效液相色谱-串联质谱(UPLC- MS/MS)测定植物源调味料中氯虫苯甲酰胺残留量的分析方法。
1 材料与方法
1.1 材料
供试样品:从海口市场抽取葱、大蒜、干辣椒、黑胡椒、白胡椒等样品各20个,其中葱为新鲜植株可食部分,大蒜为去皮后可食部分,干辣椒、黑胡椒、白胡椒均为干样;从海南、安徽、湖南等产地抽取姜与姜茎样品各20个,其在田间施用5%氯虫苯甲酰胺悬浮剂7 d后采集样品。姜茎和葱样品切断后用打浆机粉碎,姜和大蒜样品直接使用打浆机粉碎,干辣椒、黑胡椒和白胡椒使用粉碎机进行制备,得到待测样品。
实验仪器包括超高压液相色谱系统(美国Waters公司),AB SCIEX API4000+质谱系统(美国AB SCIEX公司),ACQUITY UPLC?BEH C18色谱柱(100 mm×2.1 mm×1.7 ?m,美国Waters公司),高速匀浆机(德国IKA公司),微型旋涡混合仪(上海沪西分析仪器厂)。
本实验用水均为超纯水,由美国Millipore超纯水系统制备;乙腈、甲醇(色谱纯,美国Fisher试剂公司);氯化钠(分析纯,广州化学试剂厂);PSA吸附剂(N-丙基乙二胺,分析纯,Biocomma公司);1000 ?g/mL氯虫苯甲酰胺标准溶液,购自农业农村部环境质量监督检验测试中心(天津)。
1.2 方法
1.2.1 样品的前处理 实验使用改进的QuEC hE RS方法对氯虫苯甲酰胺进行提取和净化。准确称取调味料样品10.0 g 样品至50 mL离心管中,为提高提取效率,干样使用超纯水进行润湿,其中姜茎样品加入2 mL超纯水、黑胡椒和白胡椒样品加入10 mL超纯水、干辣椒样品加入12 mL超纯水,然后再加入20 mL乙腈进行提取;高速匀浆2 min,加入1.5 g NaCl,蜗旋剧烈振荡1 min后,以4000 r/min的转速离心5 min,分取5 mL上清液转入10 mL的PSA(150 mg)净化管中,同时加入C18吸附剂,涡旋剧烈振荡1 min,再以4000 r/min的转速离心5 min,取上清液2.0 mL经0.22 ?m微孔滤膜过滤,使用UPLC-MS/MS进行测定。
1.2.2 仪器分析条件 色谱条件:ACQUITY U PL C?BEH C18色谱柱,柱温35 ℃,进样量5 ?L,流速为0.25 mL/min;流动相A为0.2%甲酸水,B为乙腈;梯度洗脱,洗脱程序:0~1 min,90% A;1.0~ 3.5 min,10% A;3.5~5.0 min,90% A。
质谱条件:电喷雾正离子(ESI+)扫描,喷雾电压4500 V,毛细管温度600 ℃;多反应监测(MRM)模式。分别选择484.1/285.9和484.1/ 452.7作为定量和定性离子,去簇电压为82.1 V,碰撞电压分别为20.0、22.0 V。
2 结果与分析
2.1 前处理方法的选择
传统农药残留分析时往往需要复杂的前处理步骤,样品制备成本较大,提取溶剂使用量多,净化步骤繁琐。QuEChERS方法因其分析速度快、抗干扰能力强、灵敏度高等独特的优势,已经广泛应用于农药残留实验的前处理。由于调味料样品中含有大量挥发性硫化物、酚类、醇类化合物等杂质[17-19][9],在使用QuEChERS方法进行前处理时,样品净化效果不佳。因此,在PSA净化时加入C18粉末进行协助净化,实验首先对比了加入C18粉150 mg后样品的净化效果,实验结果见图2A。C18材料的加入可以有效地去除调味料样品中复杂基质对氯虫苯甲酰胺提取率的影响,将氯虫苯甲酰胺的回收率从42%~72%提高到81%~ 103%。实验进一步优化了不同质量的C18粉末对氯虫苯甲酰胺提取率的影响,实验结果见图2B。结果表明,当C18质量从50 mg增加至150 mg时,不同调味料基质中氯虫苯甲酰胺的回收率从39%~68%提高到89%~106%,当C18质量从150 mg增加至250 mg时,氯虫苯甲酰胺的回收率无显著变化,因此,在后续的实验中添加150 mg的C18粉末进行净化。
2.2 检测条件优化
2.2.1 液相色谱条件 使用C18超高压液相色谱柱对氯虫苯甲酰胺进行测定,结果发现,氯虫苯甲酰胺在C18色谱柱上具有较好的保留性,可以满足分析的需求。实验进一步比较了乙腈水和乙腈-0.2%甲酸水2种流动相体系,结果表明,加入0.2%甲酸与乙腈-水流动相中,在改善峰型的同时还可使基线更加稳定。在正离子模式下,0.2%甲酸的加入还增加了氯虫苯甲酰胺离子化所需的H+,不仅增强了离子化效率,还提高了检测灵敏度。因此选取了乙腈-0.2%甲酸水体系作为流动相。同时优化调整了0~1.0 min、1.0~3.5 min、3.5~ 5.0 min,3个时间段流动相中乙腈和0.2%甲酸水的比例,确定了最佳的梯度洗脱条件。
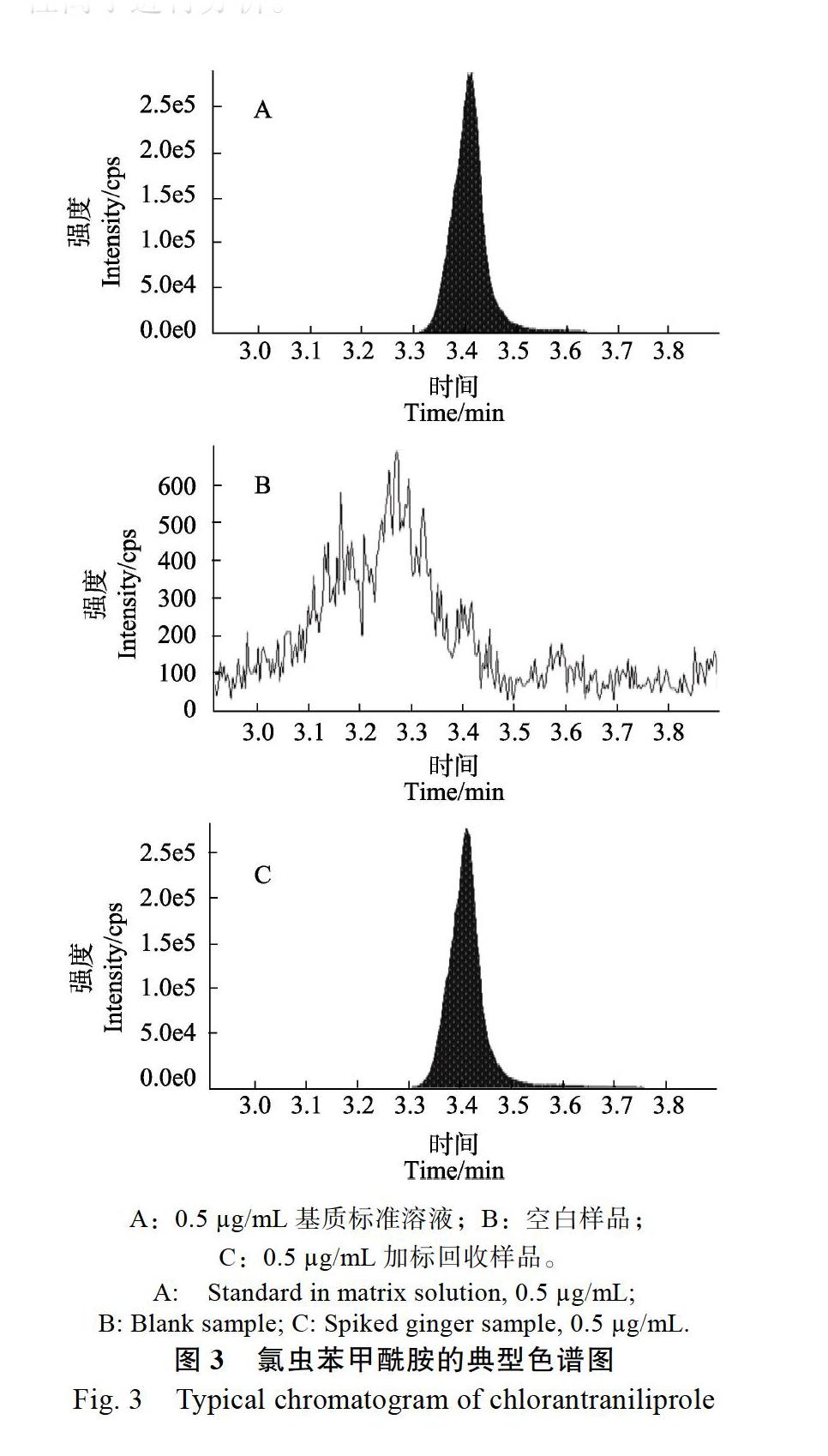
2.2.2 质谱条件优化 本方法的典型色谱图见图3,采用多反应监测模式(MRM)进行检测,MRM可以通过消除背景干扰来提高检测灵敏度。首先对0.5 ?g/mL的氯虫苯甲酰胺标准溶液注入ESI離子源中,在正离子检测方式下进行一级质谱分析,得到稳定且丰度高的母离子碎片m/z 484.1。进一步优化去簇电压后,对母离子碎片进行二级质谱分析,得到二级质谱碎片的全谱信息。优化碰撞电压,选择两个信号最强的两个碎片离子m/z 285.9、m/z 452.7离子对,分别作为定量离子和定性离子进行分析。
A:0.5 ?g/mL基质标准溶液;B:空白样品;
C:0.5 ?g/mL加标回收样品。
A: Standard in matrix solution, 0.5 ?g/mL;
B: Blank sample; C: Spiked ginger sample, 0.5 ?g/mL.
2.3 基质效应
实验分别使用姜、姜茎、葱、蒜、干辣椒、黑胡椒、白胡椒等7种调味料空白样品,按照样品提取方法对样品进行前处理,得到样品提取液,分别使用纯溶剂和样品提取液配制0.005、0.050、0.200 ?g/mL 3个浓度水平氯虫苯甲酰胺溶液,分别测定得到纯溶剂中农药的响應值(A)和样品提取液中农药响应值(B),则样品基质效应= A/B×100%。实验结果表明,经过前处理后植物源调味料样品的基质效应在91%~115%,说明本方法具有很好的净化效果,可以较好的去除样品中基质的干扰。
2.4 标准曲线及方法检出限
以浓度1000 ?g/mL的氯虫苯甲酰胺标准溶液作为母液,使用甲醇逐级稀释法分别配置0.005、0.02、0.05、0.1、0.2 ?g/mL的系列标准溶液,外标法进行定量,按所确定检测条件,以氯虫苯甲酰胺对照溶液浓度(X)为横坐标,峰面积(Y)为纵坐标,绘制标准曲线。得到回归方程Y=2795500X+22700,线性关系良好,相关系数优于0.999。以定量离子响应的3倍信噪比和10倍信噪比(S/N)分别计算检出限(LOD)和定量限(LOQ),得到氯虫苯甲酰胺的LOD和LOQ分别为0.002 mg/kg和0.005 mg/kg。其中,LOQ低于国标GB 2763-2016中所规定的最低氯虫苯甲酰胺残留限量值(0.02~20 mg/kg),满足国家标准的相关要求。
2.5 回收率及方法的精密度
以不含氯虫苯甲酰胺的植物源调味料空白样品,分别在0.005、0.05、0.2 mg/kg 3个水平进行加标回收率实验,每个浓度平行5次,相关结果见表1所示。实验结果的加标平均回收率在70%~ 108%之间,相对标准偏差(RSD)在1.9%~10%之间。参考NY/T 788-2004农药残留试验准则,实验结果加标回收率和相对标准偏差均符合农药残留测定允许范围,说明方法的准确度符合残留检测要求。
2.6 实际样品检测
利用本方法进行检测对抽取的样品进行检测,所有样品均配制基质标准进行定量分析。结果表明,研究所抽取的姜、葱、蒜、干辣椒、黑胡椒、白胡椒等样品均未检出氯虫苯甲酰胺,20个姜茎样品中,有6个样品被检出,浓度范围为0.082~0.263 mg/kg。本方法具有良好的灵敏度和重现性,可用于满足市场样品监测的需求。
3 结论
植物源调味料中含有大量硫化物、酚类、醇类化合物通常影响农药残留的定量分析,其回收率较低。传统的QuEChERS方法使用PSA作为净化材料,由于PSA材料的硅胶表面键合极性官能团,主要吸附样品中的强极性杂质,如有机酸、色素以及糖等。C18材料含有十八烷基,对非极性物质有较高的容量,其可去除油脂等非极性杂质,并对样品中硫化物等杂质具有很好的去除效果。因此,本方法同时使用PSA和C18材料进行净化,有效地去除了样品中的杂质干扰。此外,由于干辣椒、胡椒等样品含水量较低,使得氯虫苯甲酰胺的回收率偏低,因此在提取过程中加入2 mL超纯水,提高了方法的回收率,保证了方法在样品分析时的回收率在70%~108%之间,满足了分析的需求。
串联质谱具有良好的选择性和灵敏度,近年来已经发展成为农药残留分析的主要技术,但是方法在对于复杂样品分析的时候容易受到基质效应的干扰,对实验结果产生影响。本研究选择使用改进后的QuEChERS方法进行前处理,采用高效液相色谱串联质谱法检测植物源调味料中氯虫苯甲酰胺残留,重点通过PSA和C18材料对样品中的极性和非极性杂质进行净化,有效地消除了不同性质杂质对样品分析的影响,保证了方法的准确性,在测定范围内线性关系较好,平均回收率和RSD均满足检测要求,可很好的满足植物源调味料中氯虫苯甲酰胺残留量的检测分析。
参考文献
刘腾飞, 杨代凤, 邓金花, 等. 氯虫苯甲酰胺的残留降解与检测分析研究进展[J]. 中国农学通报, 2015, 31(3): 221-228.
陈小军, 费 春, 樊丽萍, 等. 氯虫苯甲酰胺在大豆植株中的内吸传导特性[J]. 中国农业科学, 2011, 44(11): 2276- 2283.
范 君, 刘腾飞, 杨代凤, 等. 农产品与环境样品中氯虫苯甲酰胺的残留动态分析[J]. 中国农学通报, 2016, 32(35): 51-57.
吴玉娥. 氯虫苯甲酰胺在水稻上的残留分析和消解动态研究[D]. 贵阳: 贵州大学, 2017.
仇贵生, 张怀江, 闫文涛, 等. 氯虫苯甲酰胺对苹果树桃小食心虫及金纹细蛾的控制作用[J]. 昆虫知识, 2010, 47(1): 134-138.
亢晓冬, 孙 霞, 沈 礼, 等. 氯虫苯甲酰胺的高效液相色谱分析方法研究[J]. 浙江化工, 2010, 41(4): 31-32,15.
钱鸣蓉, 章 虎, 吴俐勤, 等. 高效液相色谱-串联质谱法测定蔬菜中氯虫苯甲酰胺和氟虫双酰胺残留[J]. 分析化学, 2010, 38(5): 702-706.
杨 森, 吴 勇, 谢龙安. 高效液相色谱法测定大米中氯虫苯甲酰胺[J]. 南方农业, 2014, 8(12): 113-114.
马婧玮, 马 欢, 安 莉, 等. QuEChERS-高效液相色谱-串联质谱法测定主要谷物和油料作物中氯虫苯甲酰胺的残留[J]. 农药学学报, 2018, 20(1): 129-134.
陈小军, 王 萌, 范淑琴, 等. QuEChERS前处理结合HPLC-MS/MS法分析氯虫苯甲酰胺在甘蓝和土壤中的残留[J]. 中国农业科学, 2012, 45(13): 2636-2647.
李桂红, 李二虎, 张 武, 等. 超高效液相色谱串联质谱测定果蔬中氯虫苯甲酰胺残留[J]. 世界农药, 2018, 40(3): 58-61, 64.
朱 烈, 周 宏, 许敏球. 甘蓝和花菜中氯虫苯甲酰胺的残留与消解动态[J]. 浙江农业科学, 2014(8): 1244-1246.
李红红, 王彦辉, 韦 典, 等. 氯虫苯甲酰胺在甘蔗及土壤中的残留消解动态[J]. 农药学学报, 2016, 18(1): 101-106.
杨玉霞, 莫仁甫, 周其峰, 等. 氯虫苯甲酰胺在果蔗及其土壤中殘留分析方法[J]. 西南农业学报, 2016, 29(11): 2643-2647.
朱建华, 赵 莉. 液相色谱串联质谱法测定果蔬中的唑虫酰胺、氟啶虫酰胺、氯虫苯甲酰胺及氟虫双酰胺残留[J]. 分析测试学报, 2011, 30(6): 646-650.
张 云, 李今中, 李耀平, 等. 液相色谱法测定动物源性食品中氯虫苯甲酰胺和氟虫酰胺残留量[J]. 分析试验室, 2012, 31(4): 119-122.
刘 瑜, 蒋 施, 徐宜宏, 等. 气相色谱-串联质谱法测定葱、姜、蒜中120种农药残留量[J]. 化学通报, 2012, 75(12): 1132-1139.
贾 斌, 李 瑾, 冯书慧, 等. 微波处理-气相色谱法快速测定葱蒜类蔬菜中氟环唑残留[J]. 农药, 2009, 48(6): 442-444.
倪永付, 王志宏, 闫秋成, 等. 搅拌棒萃取-GC-MS/MS法检测黑蒜汁中8种有机氯农药残留[J]. 食品工业, 2016, 37(10): 294-297.
