植物乳杆菌细菌素基因plnEF的克隆与异源表达
2019-08-28阮晓莉冉军舰赵瑞香ZHUYang
阮晓莉,冉军舰,赵瑞香,*,李 刚,雷 爽,ZHU Yang
(1.河南科技学院食品学院,河南新乡 453003;2.瓦赫宁根大学研究中心,格尔德兰瓦赫宁根 5960AA)
细菌素是指某些细菌在代谢过程中产生的一类抗菌性的多肽、蛋白质或蛋白质复合物[1],能够有效抑制或杀灭致病菌和腐败菌,近缘菌种产生的细菌素会呈现出较为狭窄的抑菌圈[2]。细菌素相较于抗生素更加高效,且在实际应用中无毒副作用和耐药性,某些产生菌在发酵过程中还能起到一定的保健作用,因此将细菌素应用在食品领域中的前景可观[3-4]。目前细菌素主要应用于分子生物学领域,研制开发能够代替医疗保健剂的产品,虽然细菌素的种类繁多,但是在食品防腐剂方面应用较少[5]。迄今为止仅有一种羊毛硫细菌素Nisin被世界上六十多个国家允许使用在食品防腐方面,绝大多数的细菌素尚在试验阶段,主要是因为化学合成的细菌素成本高、性质不稳定而且天然细菌素的产量比较低,难以纯化进行商业化生产,因此通过基因重组技术以期获得大量纯度较高的细菌素并将其应用到实际当中显得尤为重要[6-7]。
细菌素分为四类:Ⅰ类为羊毛硫抗生素,Ⅱ类为含有非修饰氨基酸的多肽,Ⅲ类为大分子蛋白质,Ⅳ类为蛋白质复合物[8]。Ⅱ类细菌素被分为Ⅱa、Ⅱb、Ⅱc和Ⅱd四个亚类,其中研究较为深入的是Ⅱa类细菌素[9-10],但是其作为异源表达的对象时,最终获得的抗菌性蛋白活性差异显著,究其原因主要是Ⅱa类单肽链非修饰抗菌肽对宿主细胞产生一定的毒性作用[11],经多年来研究人员对细菌素遗传学的深入研究发现,将Ⅱb类细菌素进行异源表达可以克服毒害宿主细胞的问题[12]。乳酸菌作为世界上广泛应用的益生菌,目前对于其产生的Ⅱb类细菌素的研究相对较少,植物乳杆菌素PlnEF属于Ⅱb类双肽链非修饰细菌素,但天然的细菌素PlnEF不仅产量低而且难以分离纯化,最实用、高效率的方法就是通过基因工程来解决此类难题。
表达质粒pET含有强启动子使其具有强大的表达功能,并且本身携带6个组氨酸的His单抗标签,使融合蛋白能够得到较好地鉴定与纯化[13]。大肠杆菌(Escherichiacoli,E.coli)因其多方面的优越性多年来广泛应用于生物工程领域,其作为表达菌株具有清楚的遗传背景、培养条件简单、价格低廉且外源表达水平远高于其他宿主细胞等优点,所以常作为生物技术研究的首选表达体系[14]。早在2004年,Park等[15]将导入有植物乳杆菌(Lactobacillusplantarum)谷氨酸脱羧酶基因的大肠杆菌进行诱导表达,获得了重组谷氨酸脱羧酶蛋白的高效表达,并证实该蛋白具有活性。龚钢明等[16]将乳杆菌中亚硝酸盐还原酶基因连接质粒pET-32a(+)构建表达载体,并成功导入受体细胞大肠杆菌中,使其大量克隆后表达的蛋白达到预期的生物特性。植物乳杆菌含有产生细菌素的基因,可依据植物乳杆菌抗菌特性探究其作为生物防腐剂的潜力[17]。对于表达系统的鉴定,比较常用的方法是将工程菌菌落通过PCR扩增目的基因,确定目的基因是否成功导入质粒载体,若想进一步验证原核表达质粒是否构建成功,可提取重组质粒进行双酶切鉴定,并将鉴定结果呈阳性的重组质粒送入测序公司测序。不少研究者在此类分子生物学研究中采用了上述鉴定方法[18-20]。
本研究采用基因工程技术,在大肠杆菌BL21中导入重组质粒pET-28a-PL-plnEF构建工程菌表达抗菌性蛋白,经镍柱纯化以期获取细菌素的高效表达,对深入探究植物乳杆菌细菌素的生物特性以及开发其作为食品防腐剂提供理论基础。
1 材料与方法
1.1 材料与仪器
植物乳杆菌(Lactobacillusplantarum) 从水开菲尔分离并于实验室保藏;表达质粒pET-28a(+)、指示菌株大肠杆菌(EscherichiacoliJM109)、大肠杆菌BL21(EscherichiacoliBL21) 由河南科技学院生物技术试验室保存;SOB(Super Optimal Broth)培养基 海博生物技术有限公司;MRS(Man-Rogosa-Sharpe)、LB(Luria-Bertani)培养基 所用试剂均为国产分析纯;SanPrep柱式质粒DNA抽提试剂盒 生工生物工程股份有限公司;胶回收试剂盒 OMEGA公司;Prestained DL 15000 DNA Marker、Prestained DL 2000 DNA Marker、2×Power Taq PCR MasterMix 北京百泰克生物技术有限公司;HisTrap HP亲和色谱柱 GE Healthcare公司;基因组DNA的小量纯化试剂盒、T4 DNA Ligase Buffer、限制性核酸内切酶Xho I、Noc I、Premixed Protein Marker(Low) 日本TaKaRa公司。
VCX130超声波细胞破碎仪 美国SONICS公司;PCR扩增仪 Long Genes Instruments;ZHYW-211C恒温培养振荡器 上海智城分析仪器制造有限公司;Sigma3K15台式高速冷冻离心机 德国Sigma公司;GelX1650凝胶成像仪 上海欧翔科学仪器有限公司。
1.2 实验方法
1.2.1 重组质粒的构建及异源导入
1.2.1.1 菌株的培养及DNA提取 取超低温冰箱保存的植物乳杆菌菌种在平板中划线,挑取单菌落于MRS液体培养基中37 ℃培养24 h,按1%的接种量于培养基中培养18 h作为种子液。使用TAKARA基因组DNA的小量纯化试剂盒提取植物乳杆菌基因组DNA。
1.2.1.2 植物乳杆菌素plnEF基因扩增 根据美国国立生物技术信息中心(NCBI)中报道的plnEF(432 bp)基因片段,选择限制性核酸内切酶Xho I、Noc I作为目的基因plnEF片段5′端和3′端的內切酶位点,在pET-28a(+)质粒本身携带的His标签单抗,构建含有6个组氨酸标签的表达载体,蛋白优化表达后可得到纯度较高的细菌素。
根据目的基因序列利用prime5.0在线软件设计引物,由武汉金凯瑞生物有限公司合成,碱基序列如下:前端引物plnEF-F(Xho I):5′-ATGCCTCGAGT GGGCATAGTTAAAA-3′;后端引物plnEF-R(Noc I):5′-TATGGATTCTCAGGTTGCCGCAAAA-3′,质粒克隆位点如图1。
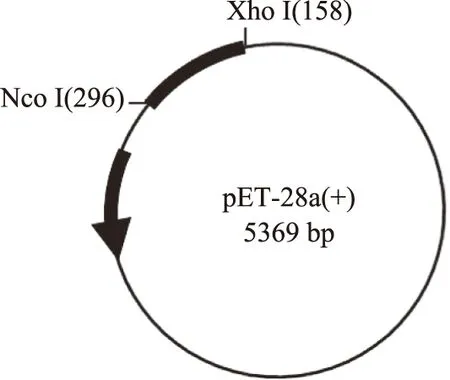
图1 质粒pET-28a(+)克隆位点图
以供体菌株植物乳杆菌中全基因组为模板,PCR扩增目的基因,随后采用1%琼脂糖凝胶电泳验证PCR产物[21]。反应条件:95 ℃预变性5 min,95 ℃变性、55 ℃退火、72 ℃延伸各30 s,30个循环[22]。
1.2.1.3 纯化目的基因 PCR扩增产物中含有少量引物二聚体,需要将DNA进行胶回收[23],目的在于分离这些基因片段,将凝胶中目的基因所在的区域切胶回收其中的基因片段,以达到纯化目的基因的目的。胶回收后的样品用1%的琼脂糖凝胶验证目的基因是否已回收到及其片段大小是否正确。
1.2.1.4 构建表达载体 将纯化后的目的基因与提取出的pET-28a(+)质粒分别用限制性核酸内切酶Xho I、Noc I在保温仪中37 ℃双酶切3 h并验证[24]。酶切体系:10×QuickCut Green Buffer 5 μL;限制性核酸内切酶Noc I、Xho I各1 μL;质粒pET-28a(+)15 μL;加双蒸水至50 μL。双酶切后1%琼脂糖凝胶电泳验证分子量大小。将目的基因利用T4 DNA连接酶连接至质粒并命名为pET-28a-PL-plnEF。
利用电穿孔仪选择Bacteria模式Ecl2(2.49 kv,5.9 ms),将表达载体pET-28a-PL-plnEF导入感受态大肠杆菌BL21并迅速加入1 mL SOB培养基,培养1 h后菌液高速离心余留150 μL培养基用于重悬菌体,涂于预热好的含Kana抗生素的LB平板[25]。
1.2.1.5 BL21-pET-28a-PL-plnEF重组菌株鉴定 12 h后培养完成的LB平板上出现白色菌落,为了确认目的片段已成功转化到宿主细胞BL21中,需要选择阳性单菌进行PCR鉴定,此时所用引物无限制性酶切位点,反应条件参照1.2.1.2。
1.2.2 融合蛋白表达及验证
1.2.2.1 融合蛋白的表达及提取 为验证目的蛋白是由目的基因plnEF转录而成的,需要将空白质粒电转化大肠杆菌作为对照。同时对成功构建的工程菌BL21-pET-28a-PL-plnEF和对照组BL21-pET-28a诱导表达[23-24]。即在500 mL培养基中按1∶1000加入50 mg/mL Kana,分别将两种工程菌以1%的比例转接至培养基,继续37 ℃振荡培养5 h后加入IPTG,IPTG作为诱导剂能够启动基因发生转录,在30 ℃条件下诱导反应12 h。若菌量比较少可以将菌液离心浓缩至两管弃除上清液,按比例1∶20加入ddH2O重悬菌体,超声破法处理菌液(超声条件:300 W,时间6 min,超声破碎3 s,停歇4 s,50个循环),取上清液再次离心用0.22 μm滤膜去除杂质,滤液收集用于SDS-PAGE蛋白验证。
1.2.2.2 融合蛋白的亲和纯化及鉴定 将1 mL HisTrap HP纯化介质装柱,上方安装0.22 μm的滤膜,首先用注射器取1 mL的蒸馏水缓慢流洗镍柱10遍,其次用浓度为10 mmol/L的咪唑(imidazole buffer with saline,IBS)缓冲溶液冲洗十遍以上,以便将柱子中容易挥发的杂质冲洗出来以达到平衡柱子的目的。将工程菌表达后得到的粗提液蛋白上样于His Trap HP 2至3次,分别用1 mL 10、20、50、100、200、300、400、500 mmol/L咪唑洗脱蛋白,由预实验可知,50 mmol/L咪唑可洗脱目的融合蛋白,对照组所得蛋白用50 mmol/L咪唑。SDS-PAGE(15%分离胶、5%浓缩胶)电泳鉴定。
1.2.3 抑菌性试验 为确定目的抗菌性蛋白的洗脱梯度及抑菌效力,以大肠杆菌JM109为指示菌株进行抑菌性试验。牛津杯法在LB平板中打孔,分别取工程菌BL21-pET-28a-PL-plnEF和对照组工程菌BL21-pET-28a预留的培养基上清液以及不同浓度的咪唑缓冲液冲洗出的蛋白滤液各100 μL加至孔中,正置于恒温培养箱中37 ℃培养,每组做3个平行试验。
为确定目的抗菌性蛋白的洗脱梯度及抑菌效力,以大肠杆菌JM109为指示菌株进行抑菌性试验。牛津杯法在LB平板中打孔,分别取工程菌BL21-pET-28a-PL-plnEF预留的培养基上清液、不同浓度的咪唑缓冲液冲洗出的蛋白滤液以及对照组工程菌BL21-pET-28a用50 mmol/L咪唑洗脱的蛋白滤液各100 μL加至孔中,正置于恒温培养箱中37 ℃培养,每组做3个平行试验。
1.3 数据处理
每组试验进行三次重复,试验结果用平均值±标准偏差显示,采用SPSS(18.0)软件处理数据。
2 结果与分析
2.1 重组质粒的构建及异源导入
2.1.1 胶回收目的基因验证 如图2所示,PCR扩增产物经胶回收后已完全去除引物二聚体形成的非目的条带,达到纯化的目的。

图2 胶回收目的基因验证结果
2.1.2 双酶切目的基因、pET-28a(+)以及酶连产物验证 双酶切目的基因仅切掉6个保护碱基,验证结果如图3。一般试剂盒提取出的质粒呈高度折叠状态,需要经过酶切后再进行电泳,以确定质粒的实际大小。已知表达质粒分子量为5369 bp,质粒经限制性核酸内切酶Xho I、Noc I切掉132 bp,琼脂糖电泳结果如图4显示,质粒分子量大小无明显变化,在大约5000 bp的位置出现唯一且明显的条带,证明质粒双酶切成功。
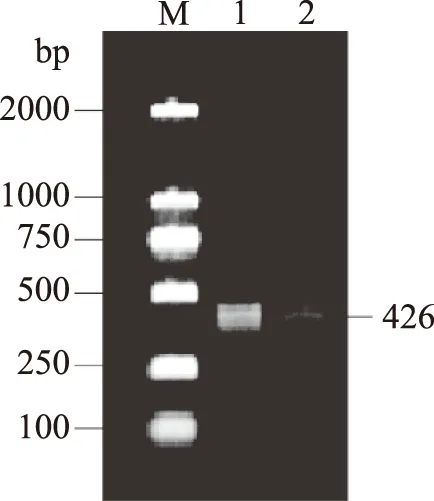
图3 双酶切目的基因验证结果

图4 双酶切质粒pET-28a(+)验证结果
连接产物验证结果如图5,可明显看到近6000 bp有一条带,与预期酶连产物分子大小相符。

图5 酶连产物验证结果
2.1.3 原核重组表达质粒的鉴定 重组质粒菌落PCR扩增后经琼脂糖凝胶电泳验证,结果如图6所示,出现唯一且较为明显的目标条带,条带位置预期值与目的基因plnEF426 bp大小相符,表明重组质粒PCR扩增效果良好,初步证明目的基因plnEF已整合到质粒pET-28a(+)中[26-27],测序结果与plnEF序列一致。
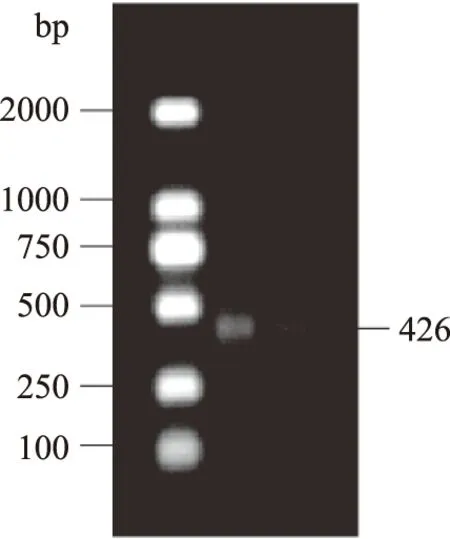
图6 菌落PCR鉴定
2.2 目的融合蛋白的纯化
工程菌菌液采用超声破碎法破碎细胞,释放目的融合蛋白,高速离心获得粗提液蛋白。经不同浓度的咪唑镍柱纯化,收集洗脱蛋白进行SDS-PAGE电泳鉴定[28-29]。染色3 h脱色后,50 mmol/L咪唑溶液洗脱后呈现出一条分子量在14.3~20.1 kDa之间的蛋白条带,基本与理论融合蛋白分子量15.6 kDa大小相符(图7)。空白对照在50 mmol/L咪唑洗脱时没有此蛋白条带,表明该融合蛋白应为目的蛋白,且能够在50 mmol/L咪唑浓度下得到有效溶解,纯化效果较好。

图7 BL21-pET28a-PL-plnEF及对照组诱导表达结果
2.3 抑菌性试验
如图8所示,LB平板上出现明显大小不同的抑菌圈,指示菌株浓度一定,抑菌圈大小与抗菌蛋白的抑菌效力有关,依据平行实验,平板中1至11抑菌圈平均直径如表1所示。其中3号即50 mmol/L咪唑洗脱的目的融合蛋白抑菌效果最为明显,说明目的融合蛋白具有抗菌作用。由抑菌圈11可明显看出50 mmol/L咪唑洗脱下的对照组工程菌蛋白没有抑菌能力,且通过SDS-PAGE电泳结果泳道3和10对比得出,对照组工程菌BL21-pET-28a没有产生50 mmol/L浓度的咪唑能够有效洗脱的蛋白。结合SDS-PAGE电泳蛋白表达结果以及抑菌性试验分析可知,确定50 mmol/L的咪唑洗脱的融合蛋白,即为目的基因plnEF所表达的目的蛋白,且该蛋白具有较高的抑菌活性[30]。
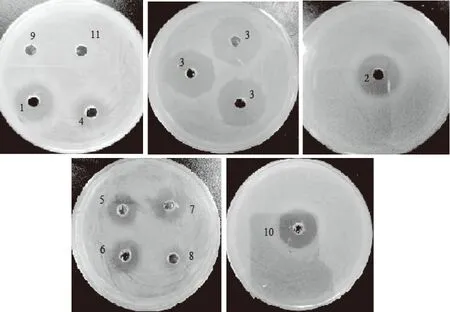
图8 BL21-pET-28a-PL-plnEF蛋白及对照组蛋白抑菌性图
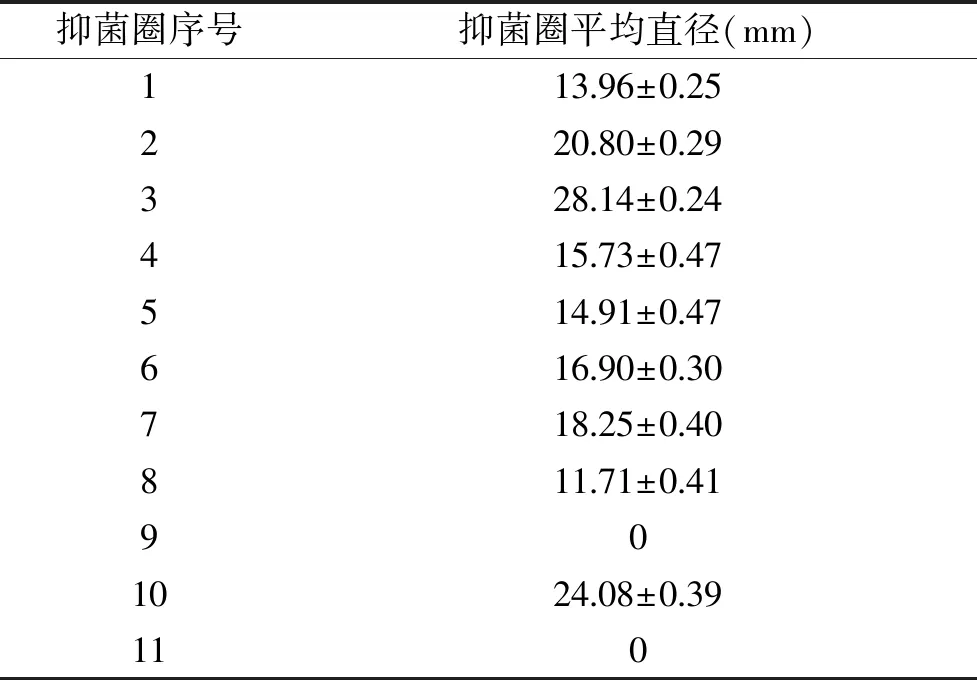
表1 抑菌试验中各抑菌圈直径平均值
3 结论
本实验中菌落直接PCR鉴定后将重组质粒测序,不仅能降低实验成本,大大缩短试验时间,而且减少了实验操作中各种因试剂残留对分子鉴定带来的不利影响。在本实验中工程菌按1%的接种量仅培养5 h进行IPTG诱导,蛋白获得高效表达,但不同诱导条件对重组融合蛋白的表达有很大影响。在本试验中若想使重组融合蛋白最大程度的表达,需要对诱导剂在不同诱导时间、诱导环境、诱导浓度等条件下做进一步的研究。
理论目的蛋白条带为15.6 kDa,与SDS-PAGE电泳鉴定结果相符。通过抑菌试验结果可以明显观察到浓度为50 mmol/L的咪唑能有效洗脱该工程菌所表达的蛋白且该蛋白抑菌作用较强,工程菌发酵上清液具有明显抑菌效果,说明发酵上清液中同样含有该目的抗菌蛋白,依据抑菌圈直径大小表明,50 mmol/L咪唑洗脱下来的目的蛋白与其发酵上清液相比抗菌效力更强,本试验中的目的融合蛋白是以可溶性表达的形式在工程菌中进行高效表达,表明该蛋白对宿主细胞BL21没有毒害作用。基于抑菌试验对该蛋白的抑菌性检测,植物乳杆菌plnEF基因片段编码的多肽能够在原核细胞中正确表达,能够作为潜在的食品生物防腐剂,可在不同条件下研究其抑菌活性和特性,将成为进一步研究的方向。
