托莫西汀合成工艺改进
2019-08-28张秋月尤启冬杨金鹏
张秋月,尤启冬*,杨金鹏
(1中国药科大学江苏省药物设计与优化重点实验室,南京210009;2南京卡文迪许生物工程技术有限公司,南京210000)
托莫西汀(atomoxetine,1)是一种高度选择性去甲肾上腺素再摄取抑制剂(norepinephrine reuptake inhibitor,NRI),通过选择性地与神经突触前膜上的去甲肾上腺素(noradrenaline,NE)再摄取转运体结合,抑制NE再摄取,而与其他神经递质亲和力极低。托莫西汀是第1个用于治疗注意缺陷多动障碍(attention deficit hyperactivity disorder,ADHD)的非兴奋性药物,目前已经在美国、澳大利亚、加拿大、英国、中国等多个国家上市并应用[1]。在2016版《中国注意缺陷多动障碍防治指南》中,托莫西汀被推荐为一线治疗药物之一,其治疗效果显著,不良反应的发生率低[2]。
文献报道的托莫西汀的合成方法主要有以下4种。
路线1[3]以 3-(二甲基氨基)-苯丙酮-1为原料,经硼烷还原、氯代、Williamson醚化和去甲基化4步反应得到外消旋托莫西汀,且中间体氨基醇的氯代会产生脱水副产物,而且在最后一步反应采用了毒性较大的去甲基化试剂(BrCN)进行N-去甲基化反应。
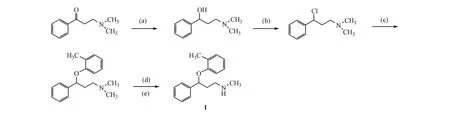
Scheme 1 Synthesis of tomoxetine by Route 1Reagents and conditions:(a)diborane in tetrahydrofuran,r.t.,overnight;(b)HCl,SOCl2,reflux,5 h;(c)o-cresol,NaOH,methanol,reflux,5 days;(d)BrCN,benzene,N2,r.t.,overnight;(e)KOH,ethylelene glycol,reflux,20 h
路线2[4]采用酶催化的不对称合成,以苯甲酰基乙酸乙酯为原料,经酶催化还原、甲胺缩合、四氢铝锂还原、Boc保护仲氨基、Mitsunobu反应及脱Boc保护基6步反应得到托莫西汀,但反应过程中涉及复杂酶催化操作和无水、无氧条件下的LiAlH4还原,反应路线较长,工业化制备难度较大。
路线 3[5]和路线 4[6]采用苯乙酮为原料,经过6步反应得到托莫西汀,反应路线较长,且均需通过化学拆分的方法得到关键中间体或目标产物,收率较低(路线3总收率9.3%、路线4总收率分别为10.5%)。

Scheme 4 Synthesis of tomoxetine by Route 4Reagents and conditions:(a)NH2 CH3·HCl,paraformaldehyde,ethanol,65°C,6 h;(b)K2 CO3,NaBH4,water/methanol,0 5°C,36 h;(c)(Boc)2O,CH2 Cl2,35°C,1 h;(d)o-cresol,DIAD,triphenylphosphine,toluene,r.t.,24 h;(e)HCl,EA,r.t.,6 h;(f)L-(-)-DPTTA,EA,55°C,2 h;(g)NH3(gas),ethanol,30 40°C,4 h
本研究在分析文献方法的基础上,对托莫西汀的制备工艺进行改进,采用CBS(Corey-Bakshi-Shibata)不对称还原法制备托莫西汀。CBS还原是一种常用的不对称还原法,具有催化剂用量少,操作简便的特点[7]。本研究以 3-氯-1-苯基丙-1-酮(2)为起始原料,通过CBS不对称还原得到高光学纯的(S)-3-氯苯丙醇,再通过Mitsonubu反应,翻转构型,得到(R)-1-(3-氯-1-苯基丙氧基)-2-甲基苯(化合物4);最后甲胺化得到托莫西汀,合成路线见图5。第三步甲胺化反应,文献[8-9]采用130℃封管反应,收率为60%,本研究采用常压甲胺化,收率41%,易于工业化。改进后的工艺操作简化,反应条件温和,为托莫西汀的制备提供了一种新的方法。

Scheme 5 Synthetic route of tomoxetineReagents and conditions:(a)NaBH4,SnCl2,(R)-(+)-alpha,alpha-diphenyl-2-pyrrolidinemethanol,THF,reflux,1 h;(b)o-cresol,DIAD,triphenylphosphine,THF,0 5°C,2 h;(c)25%30%methylamine in aqueous solution,ethanol,reflux,48 h
1 实验部分
1.1 仪器及试剂
LC-10ATVP高效液相色谱仪(日本岛津公司);Micromass LCT液质联用仪(美国 Waters公司)及Agilent 1100SL型离子阱质谱仪(美国安捷伦公司);AV-400型核磁共振仪(瑞士 Bruber公司,TMS为内标);WZZ-3型自动旋光仪(上海精密科学仪器有限公司);YRT-3型熔点仪(天津大学精密仪器厂)。
3-氯-1-苯基丙-1-酮(2)(上海嵩爱化工科技有限公司,含量:≥98.3%,批号:20180825-SY6);R-(+)-α,α-二苯基脯氨醇(上海嵩爱化工科技有限公司,含量:99.4%,批号:20180911-SY4);甲胺水溶液(安徽泽钜化工有限公司,含量:28.2%,批号:AHZJ-2018092403);所用溶剂均为工业级(南京润升石化有限公司);其他所用试剂均为市售分析纯。
1.2 实验步骤
1.2.1 (S)-3-氯-1-苯基丙醇(3)的制备 向二水合氯化亚锡(46.8 g,0.21 mol)中加入甲苯100 mL,在120℃下回流分水(约分出水7.5 mL),保持温度,常压蒸出甲苯,降温至15℃后,加入四氢呋喃200 mL,配制氯化亚锡的四氢呋喃溶液备用。
向1 L四颈瓶中加入硼氢化钠(33.7 g,0.89 mol)和四氢呋喃 200 mL,N2保护下,控温 15~23℃,滴加氯化亚锡的四氢呋喃溶液;滴完后,控温15℃搅拌1 h;N2气流下,分批加入R-(+)-α,α-二苯基脯氨醇(7.5 g,0.03mol);逐渐升温,回流30 min;TLC检测R-(+)-α,α-二苯基脯氨醇反应完毕(展开剂:正己烷-乙酸乙酯,5∶1),催化体系已形成后,缓慢滴加 3-氯-1-苯基丙-1-酮(2)溶液(50.0 g,0.30 mol,溶于干燥四氢呋喃 25 mL中);回流反应1 h,TLC检测3-氯-1-苯基丙-1-酮反应完毕,冰水浴降温,并滴加水淬灭;抽滤反应液,滤液减压浓缩;加入1 mol/L盐酸100 mL和乙酸乙酯100 mL,搅拌30 min,萃取分液,水相用乙酸乙酯30 mL再次萃取,合并有机相,无水硫酸钠干燥,抽滤;滤饼用乙酸乙酯淋洗;滤液减压浓缩;残留物用硅胶柱色谱分离(洗脱液:正己烷-乙酸乙酯,10∶1),得白色絮状固体(50.6 g,100%,):mp:50.7~51.9℃(文献值56~57℃[10]),HPLC法检测纯度为93.08%[面积归一化法,色谱柱:Phenomenex Luna C18(5μm,250 mm×4.6 mm);流动相:0.1%磷酸水溶液-甲醇-乙腈(体积比40∶30∶30);流速:1.0 mL/min;检测波长:210 nm;柱温:室温],MS(m/z):188.084 57[M+NH4]+。
1H NMR(400 MHz,CDCl3)δ:7.28~7.37(5H,m,Ar-H),4.92~4.95(1H,dd,J1=8.4 Hz,J2=4.8 Hz,CH),3.70~3.77(1H,m,CH2),3.53~3.58(1H,m,CH2),2.19~2.28(1H,m,CH2),2.05~2.12(2H,m,CH2,-OH)。
1.2.2 (R)-1-(3-氯-1-苯基丙氧基)-2-甲基苯(4)的制备 向100mL四颈瓶中加入化合物3(6.0 g,0.04 mol)、邻甲酚(4.6 g,0.04 mmol)、三苯基膦(11.1 g,0.04 mmol)和四氢呋喃 75 mL;N2保护,控温0~5℃,滴加偶氮二甲酸二异丙酯(DIAD,8.5 g,0.04 mmol),滴加完毕,在 0~5℃下反应2 h。TLC检测3反应完毕,45℃浓缩反应液,得粗品橙黄色油状物,用硅胶柱色谱分离(洗脱剂:正己烷),得到无色油状物(4.6 g,63%):纯度96.87%(HPLC归一法,方法同上),MS(m/z):261[M+H]+,283[M+Na]+。
1H NMR(400 MHz,CDCl3)δ:7.31~7.37(4H,m,Ar-H),7.26~7.28(1H,m,Ar-H),7.11~7.13(1H,d,J=7.2 Hz,Ar-H),6.94~6.99(1H,m,Ar-H),6.77~6.81(1H,m,Ar-H),6.62~6.64(1H,d,J=8.0 Hz,Ar-H),5.37~5.40(1H,dd,J1=8.4 Hz,J2=4.4 Hz,CH),3.78~3.84(1H,m,CH2),3.560~3.66(1H,m,CH2),2.46~2.53(1H,m,CH2),2.31(3H,s,CH3),2.20~2.28(1H,m,CH2)。
1.2.3 (R)-N-甲基-3-苯基-3-(邻甲苯氧基)丙胺(托莫西汀,1)的制备 80℃、密闭气球微压下,将化合物4的乙醇溶液(5.0 g,0.02 mol,溶于乙醇35 mL中)缓慢滴加至25%~30%甲胺水溶液(22.2 g,0.19 mol)中,回流 48 h。反应液冷却至室温,45℃浓缩除去乙醇,加入二氯甲烷萃取(25 mL×2),合并有机层,用无水硫酸钠干燥,过滤,滤液减压浓缩,残余物用硅胶柱色谱分离(二氯甲烷-甲醇体,50∶1),得橙黄色固体 3.78 g,加入乙酸乙酯10 mL打浆,得类白色固体粉末(2.0 g,41%):mp 158.7~159.8℃,[α]22D-43.8°(c1.0,MeOH,文献值 -44°[8]),纯度 99.08%(HPLC归一法,方法同上),MS(m/z):256[M+H]+。
1H NMR(CDCl3,400 MHz)δ:7.29~7.35(4H,m,Ar-H),7.23~7.26(1H,m,Ar-H),7.10~7.12(1H,d,J=7.2 Hz,Ar-H),6.93~6.97(1H,t,Ar-H),6.75~6.79(1H,t,Ar-H),6.59~6.62(1H,d,J=8.0 Hz,Ar-H),5.24~5.27(1H,dd,J1=8.4 Hz,J2=4.4 Hz,CH),2.75~2.79(2H,m,CH2),2.42(3H,s,CH3),2.32(3H,s,CH3),2.1~2.22(1H,m,CH2),1.20~2.07(1H,m,CH2),1.46(1H,s,NH)。
文献值[11]:1H NMR(free base,CDCl3)7.29~7.22(m,4H),7.18~7.14(m,1H),7.04~7.03(d,1H),6.87(t,1H),6.69(t,1H),6.53(d,1H),5.19(dd,1H),2.73~2.69(m,2H),2.35(s,3H,NCH3),2.24(s,3H,CH3),2.17~2.09(m,1H),2.00~1.95(m,1H),1.64(s,NH)。
2 结果与讨论
本研究以3-氯-1-苯基丙-1-酮(2)作为合成起始原料,经过CBS(Corey-Bakshi-Shibata)不对称还原、Mitsunobu反应和缩合3步反应得到托莫西汀,经高效液相检测纯度为99.08%,测得比旋度数-43.8°(c1.0,MeOH),与文献值 -44°(c1.0,MeOH)[8]基本一致。经改进后,工艺路线明显缩短,反应条件温和,而且避免了价格贵和高毒性试剂的使用,为托莫西汀的制备提供了一种新的方法。
