高分子前药的研究进展
2019-08-28韩天娇胡玉玺付宏征
韩天娇,胡玉玺,付宏征*
(1北京大学药学院 天然药物及仿生药物国家重点实验室,北京100191;2国家药品监督管理局药品审评中心,北京100022)
随着对人类致病机制的不断认识,潜在的靶点已基本被全面开发,每一个疾病领域新靶点的发现会带来该疾病治疗手段的突破性革新。新靶点的开发受到人类认知水平、技术表征水平等多种因素的限制,近年所开发出的全新药物相对较少。对已上市药物的二次开发,包括利用前药修饰、制备微球等特殊制剂已成为研发热点。
前药是指药物经过化学结构修饰后得到的在体外无活性或活性较小的化合物,在体内代谢释放出母体生物活性物质的药物[1]。纳米级的特殊制剂可有效改善药物的药代动力学行为,延长循环时间,同时可以利用载体的各种响应方式,实现药物的靶向输送和控制释放[2-4]。尽管两种策略具有各自的优势,但自身存在的一些缺陷限制了其临床应用。小分子前药设计存在血液中清除速率快和降解早的问题[5];传统的微纳米尺度的特殊制剂为物理载药,存在载药率低、稳定性差、药物在体循环过程中容易泄漏等缺点。利用高分子载体修饰的药物,由于高分子载体具有一定尺寸,同时自身的疏水、亲水特性可在与药物键合后,自组装成外部亲水内部疏水的纳米级药物。高分子前药可以很好的将前药修饰和纳米给药技术整合成为一种更为高效的药物修饰手段,同时将两者的劣势降低。目前,各疾病治疗领域均出现了较多高分子前药修饰的例子,尤其是抗肿瘤药领域,出现了多类型不同机制的高分子前药[6-9]。本综述根据高分子前药在体内的释药机制,主要从被动靶向、主动靶向、触发释药以及协同给药等方面进行综述,同时探讨该类药物研发过程中存在的潜在风险。
1 被动靶向前体给药系统
基于高分子前药的被动靶向给药系统主要是根据肿瘤的病理、生理特征开发出来的给药技术,特别是利用高渗透性和滞留(EPR)效应促进药物在肿瘤细胞的累积[10]。EPR效应产生的主要原因是肿瘤部位较正常组织有显著的区别,其血管丰富、血管壁间隙较宽、淋巴循环系统缺失。可供担载药物的高分子化合物种类繁多,但需满足如下条件:具有活性官能团、生物相容性好、无毒及免疫原性、可在体内降解或清除等。最常用的聚合物包括聚乙二醇(PEG)、聚N-(2-羟丙基)甲基丙烯酰胺(HPMA)、聚乳酸(PLA)、聚氨基酸、多聚糖,等。单一结构单元的高分子在载药量、药物传递效率等方面难以满足需求,利用特殊的聚合手段,合成含有不同结构链段的高分子化合物,是近年来研究比较多的高分子载体[11]。根据骨架结构,用于合成高分子前药最常用的聚合物主要包括3类:(1)嵌段共聚物;(2)树枝状聚合物;(3)梳状聚合物。这些聚合物共同的特点是在聚合物的骨架上存在大量的、数量可调控的活性官能团,可与抗肿瘤药物的活性官能团反应,在实现较高的载药量的同时也可以调节载药量,另外不同结构链段的理化性质的差异可以赋予高分子前药很多独特的性质。如PEG与疏水性多官能团的高分子化合组成嵌段共聚物后再与药物耦合,此时药物分子被聚集在疏水中心,能够得到很好的保护,同时PEG分子链在外层可以提供长循环所需要的水化层,避免被网状内皮系统快速消除,起到“隐身”的作用[12]。
如表1所示,多种高分子前药已经进入了Ⅱ期临床研究。这些药物在降低毒性,提高靶向性等方面都表现出了一定的优势,如SN38(7-乙基-10-羟基喜树碱)是伊立替康的活性代谢产物,具有严重的细胞毒性,由于其水溶性低,不能经静脉注射给药。由Enzon公司开发的利用多臂PEG与4个SN38成键,可以实现高载药量和良好的水溶性,具有很好的肿瘤组织分布特征、良好的耐受性,抗肿瘤活性明显增强,目前正在美国、日本同时开展Ⅱ期临床研究[13]。
由于肿瘤细胞的异质性和结构的复杂性,单纯依靠EPR效应实现的被动靶向作用不能有效地将活性药物分子运输到肿瘤细胞内部,同时被动靶向药物会在具有孔窗的内皮细胞上累积(如肝、脾),细胞毒性药物的副作用不能得到充分的控制。因此,开发靶向性更强、治疗效果更好的新一代药物传输体系具有重要意义。

表1 处于Ⅱ期临床试验阶段的高分子前药
2 主动靶向前体给药系统
主动靶向前体给药系统通过将高亲和力的靶向配体连接到高分子前药上,当药物富集在肿瘤组织周围后,配体可以识别靶细胞,促进药物向细胞内的转运,使靶细胞内的药物浓度增大,提高药物治疗效果。靶向递送应保证配体与其同源受体的高特异性,配体以特定的方式结合到靶细胞上,同时最小化结合于健康细胞[14]。
主动靶向高分子前药同样需要长时间的药物循环,应避免药物与血清蛋白或免疫系统发生不必要的相互作用,防止被过早的清除[15]。因此,需要优化药物表面靶向配体的密度,避免被网状内皮系统(RES)识别,保持其“隐身”特性[16]。同时,配体的密度也可以影响肿瘤细胞对药物的内吞效率,甚至可以改变药物进入肿瘤细胞的机制[17-18]。
目前,靶向抗肿瘤前药研究较多的靶点主要有转铁蛋白受体、叶酸受体、表皮生长因子受体等,根据这些靶点展开了大量的研究工作。HPMA共聚物-阿霉素-半乳糖偶合物(PK2)是其中比较突出的品种。该药物根据肝实质细胞存在大量半乳糖受体,将半乳糖配基引入到偶合物中,通过半乳糖受体介导的胞吞作用进入细胞,从而达到肝靶向的效果。研究者将半乳糖和阿霉素通过Gly-Phe-Leu-Gly四肽与HPMA共聚物偶联形成高分子前药,在体内经组织蛋白酶B降解而释放阿霉素。动物试验表明,在肝脏中药物浓度为其他正常组织的15~20倍,具有明显的肝靶向性,目前已进入Ⅱ期临床试验[19]。
3 触发释药前体给药系统
由于高分子前药键合药物的化学键相对比较稳定,因此很多高分子键合药往往存在体内药物释放过慢的问题,影响药物的治疗效果。为了解决上述问题,很多研究者应用对特定刺激敏感的化学键,设计出触发释药传递系统[20-21],该系统可以使药物在血液循环系统中保持相对稳定,而达到肿瘤部位通过特定的刺激响应机制快速释放药物[22]。由于针对肿瘤特异性释放药物的性质,触发释药系统在降低药物的副作用和提高抗肿瘤活性方面具有显著优势。
高分子前药传递系统中常用的刺激响应方式分为两类(表2):内部肿瘤微环境病理生理学的变化(比如pH、酶、还原性环境等)和外部刺激信号(比如光、热和磁场等)。
3.1 pH敏感高分子前药
在常用的刺激触发药物释放策略中,pH响应是近些年来最受关注的给药手段[23]。pH响应的高分子前药具有酸触发释放药物的特点,在系统循环的正常pH范围内,可以共价结合抗肿瘤药物,在偏酸的肿瘤组织(pH 6.5~7.0)和肿瘤细胞内(pH 5.0~6.5)中迅速释放药物[24]。酸触发药物释放可以通过应用pH敏感的可生物降解的化学键实现,主要包括缩醛键[25]、腙键[26-32]和硅醚键[33]。如,缩醛键连接的紫杉醇和聚乙二醇-聚丙烯酸嵌段共聚物(PEG-PAA)前药在酸性的内吞体/溶酶体中药物快速释放[34]。体外释放实验显示紫杉醇的释放行为有明显的pH依赖性,在pH分别为5.0、6.0和7.4时溶出度分别为86.9%、66.4%和29.0%。
3.2 还原响应高分子前药
肿瘤细胞微环境的另一个特点是含有比正常细胞高数倍的还原剂,利用这一特点开发出了还原响应高分子前药。该给药系统中研究比较多的是利用二硫键来桥接药物和聚合物,在高浓度谷胱甘肽(GSH)的细胞内环境中实现还原响应的药物释放[35]。利用二硫键桥连的 PEG-block-dendritic polylysine-喜树碱高分子前药的疏水性可以通过改变偶联的喜树碱分子的数量进行调节,并可以影响自组装药物的纳米结构(纳米球或者纳米棒)[36]。体外释放实验显示,在磷酸盐缓冲液中,该高分子前药在5 d内都没有释放任何的喜树碱,然而在二硫苏糖醇(DTT,类似于谷胱甘肽的一种强还原剂)存在下,喜树碱能够迅速释放。
3.3 酶敏感的高分子前药
一些在肿瘤中过度表达的特定的酶对于用作抗肿瘤传递刺激物具有广泛前景。在高分子前药的设计中,组织蛋白酶B、基质金属蛋白酶-2等都可作为响应载体。其中,如今研究较多的基质金属蛋白酶-2(MMP-2)在肿瘤细胞中过度表达,它与肿瘤生长、侵袭和转移密切相关[37]。通过在PEG和紫杉醇中间引进的可被MMP-2特异剪切的八肽(GPLGIAGQ),制备MMP-2敏感的PEG2000-紫杉醇偶联物,该药物在体内和体外的抗肿瘤活性比无刺激敏感的PEG2000-紫杉醇偶联物以及游离紫杉醇都强[43]。
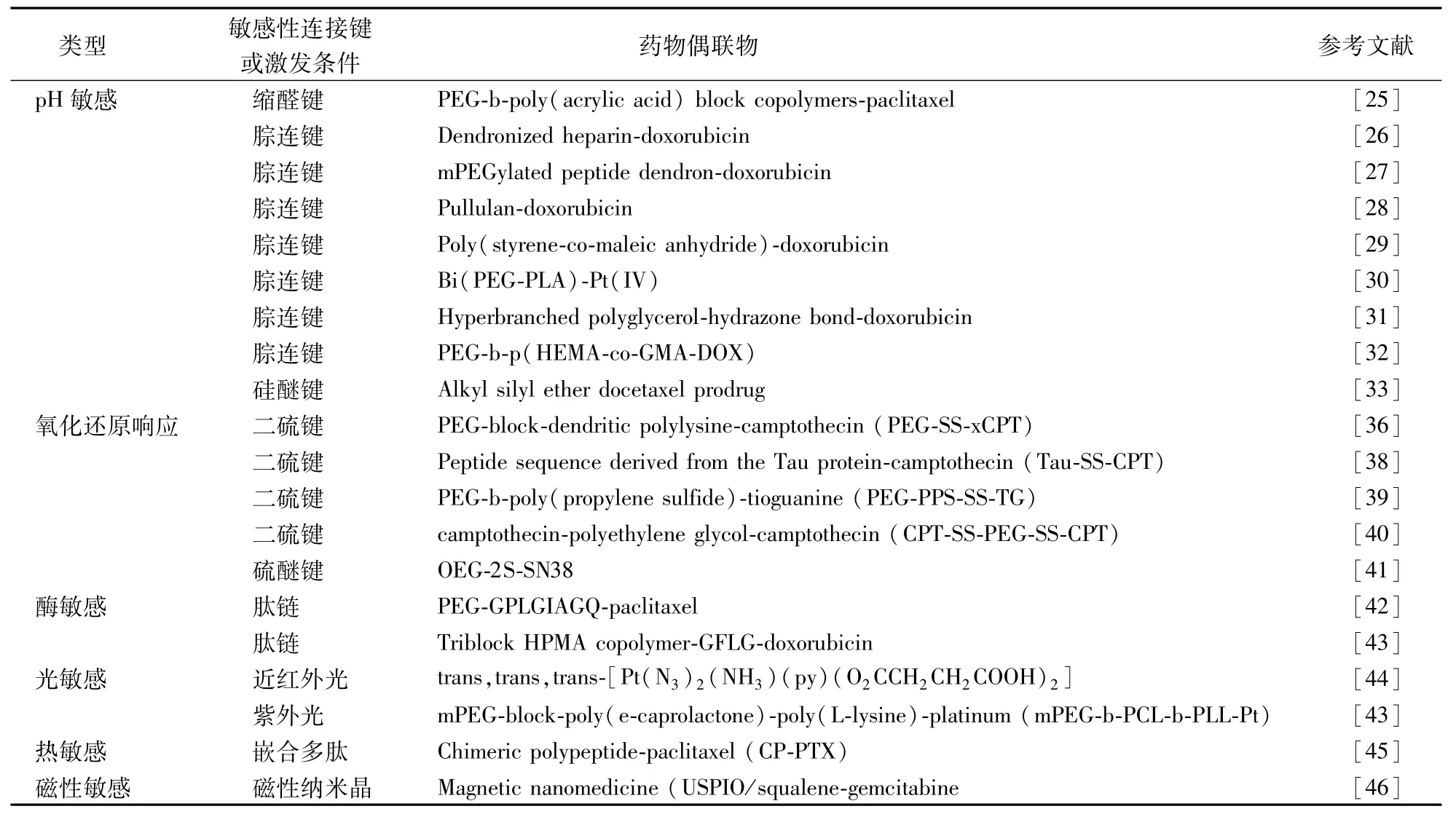
表2 高分子前药刺激响应方式
3.4 外部刺激触发释药的高分子前药
除了以上提到的根据肿瘤内部环境特点开发的高分子前药触发释药系统外,部分对外界刺激敏感的偶联药物也被设计并合成出来。其中,光、热和磁都被用来作为药物释放的刺激条件。比如,开发的光敏感铂(Ⅳ)-氧化物前药纳米组装体能够通过UVA射线的辐射实现活性Pt(Ⅱ)在肿瘤部位的选择性激发。与顺铂、奥沙利铂和游离的前药相比,光敏感铂(Ⅳ)前药纳米粒有更显著的抗卵巢癌SKOV-3细胞的作用。重要的是,光敏感铂(Ⅳ)前药纳米粒和小分子的母药物相比具有更低的系统毒性和更强的小鼠H22肝癌肿瘤生长抑制作用[47]。
4 基于高分子前药的协同给药体系
由于肿瘤的异质性和复杂性,采用单一药物治疗策略进行肿瘤治疗经常会导致耐药性的产生和肿瘤复发,多种药物联合治疗是临床上采用的主要治疗手段[48-49]。近年来,大量开创性的研究显示基于高分子前药的给药系统在药物协同传递方面具有一定的优势。
从结构上来说,基于高分子前药的联合药物治疗主要包括如下两种形式:(1)聚合物偶联两种抗肿瘤药物构成高分子前药给药系统;(2)一种药物和两亲性的聚合物偶联形成高分子前药,利用该前药的自组装特性将另一种药物包裹于其中。和单一药物化疗相比,组合药物治疗的显著优点是对肿瘤细胞可产生最大的协同化疗作用。但是,由不正确的剂量、不合适的药物比例和不准确的药物释放所引起的药物毒性,使协同给药系统的研发结果并不十分理想[50]。
和单药治疗相比,两种或者更多的化疗药物的联合应用治疗在患者身上显示更强的抑制肿瘤作用。基于铂类药物和紫杉类药物(紫杉醇、多西他赛)在临床化疗中常联合使用,通过高分子前药自组装的纳米粒药物传递系统已经应用到顺铂和多西他赛的共传递。利用疏水作用将多西他赛包裹在PLGA-PEG-COOH和PLA-Pt(Ⅳ)形成的共聚物混合体系中,形成纳米粒药物传递系统,这些纳米粒药物传递系统的表面被A10配体修饰,能够靶向前列腺癌细胞的前列腺特异性膜抗原[51]。多西他赛和铂的释放是受时间控制的,由于共价结合和非共价结合的不同,多西他赛比铂释放的速度快很多,而在紫杉类药物和铂药联用时需要紫杉类药物先作用才能起到协同效果,反之则会起到拮抗作用。因而利用高分子前药体系进行多种药物的联用时可以通过结构的巧妙设计来控制药物释放顺序,进而达到更好的协同效果。利用pH响应化学键将阿霉素与苯乙烯-马来酸酐共聚物偶联,再自组装包载双硫仑[52]。发现该载药系统具有较高的载药量及确定的药物比例,该载药系统能够保证两种药物以不同的速率释放:包载于疏水内核的双硫仑释放较快,发挥先抑制药物外排泵的活性,同时恢复耐药细胞的凋亡信号通路的作用,有效逆转肿瘤细胞对阿霉素的耐药。而通过共价偶联于聚合物上的阿霉素释放较慢,且具有pH响应性,只有在低pH条件下才能被释放出来。由于双硫仑提前抑制了药物外排泵的活性,导致阿霉素能够大量蓄积于耐药细胞中并与双硫仑产生协同作用,抑制肿瘤细胞增殖,诱导细胞凋亡,最终降低肿瘤多药耐药。动物体内研究结果显示,该药物几乎完全抑制了耐药肿瘤的生长,并显著降低阿霉素的系统毒性。
5 高分子前药研发存在的问题及解决方案
高分子前药通常由3部分组成,包含高分子载体、连接臂和活性药物。一般先通过化学反应或者高分子聚合手段将高分子链段与连接臂链接,再与活性药物键合。在设计高分子前药时,应结合药物的特点和拟修饰目的等综合考虑高分子载体和链接臂的选择。一般情况下高分子载体与药物链接后缺少有效的表征手段,因此高分子载体、连接臂和活性药物的质控体系的完整程度,以及过程控制的是否完善均对药物的批内、批间一致性有重要的影响。在常规药物研发生产管理理念的基础上建议重点考察以下环节。
5.1 高分子载体的设计
高分子载体是赋予载药新特性的重要结构单元,分子链上疏水和亲水基团的数量直接影响药物的粒子属性、稳定性、释放速度等。对高分子链的设计需结合预期达到的修饰目的综合判断结构的合理性。由于高分子载体多为含有相同结构单元的混合物,高分子载体的相对分子质量分布以及活性位点的分布情况,会影响载药数量和载药后药物粒子属性。目前,绝大多数在研高分子载体的活性位点都在链端,通过特殊的封端处理在聚合终点将活性位点引入,需重点考察活性位点引入的个数和位置。同时,活性位点的引入可能引发不同程度的高分子链聚合,导致相对分子质量分布变宽。可通过逐级沉降的方法得到相对均一的高分子载体。
5.2 连接臂的选择
实现高分子和药物的链接,重要一环为链接臂的选择是否合理,是否可满足“上得去,稳得住,下得来”的要求,即在体外环境下,可以通过简单的合成手段实现将药物和高分子载体链接,同时该化学键在体外以及未达到给药部位的时候应该相对稳定,在预期达到释药环境时,能迅速释放药物。所以在选择连接臂时,需要结合活性药物分子中存在的反应基团,以及高分子载体潜在的修饰手段进行综合判定。应对链接臂建立完善的质控体系,对与载体和药物链接过程中可能产生的副反应做充分评估。
5.3 连接活性药物的质控及表征
在与高分子载体连接之前,活性药物一般可通过有效的表征手段进行精确的结构确证,可建立其完善的质控体系。而与高分子载体链接之后,一般无法有效地确证其结构,同时也意味着无法有效表征其可能产生的杂质。因此加强链接之前的活性药物和载体的质控以及反应过程控制显得尤为重要。应充分评估活性药物的杂质水平,尤其对具有活性位点可与链接臂发生反应的杂质应该重点评估。由于杂质与连接臂的反应活性可能高于活性药物,可能大量的引入到目标药物中,可以先对连接臂上与高分子载体连接的基团利用惰性小分子基团封端,然后与药物分子模拟反应条件进行考察。由于药物与改构的连接臂反应后,可以得到结构明确的化合物,对其进行详细分析可以判断活性药物的杂质水平和拟定的反应条件是否合理。同时为了提高活性药物修饰后的纯度,降低残留的未偶联药物的高分子载体数量,一般会采取活性药物多投料的方法,使高分子载体活性反应位点尽可能地反应完全,之后采用透析等手段除去小分子药物。但是高分子载体上潜在的未反应官能团由于引入了活性团,而对药物存在一定影响,可通过滴定等手段对潜在的高分子链上未反应的活性位点进行评估,使其对药物的影响降到最低。
高分子载体修饰之后的活性药物,精确表征其结构存在一定难度。除上述应尽量加强过程控制保证得到预期的高分子前药之外,需尽可能地采用适当的表征手段对特性结构进行考察。对于修饰之后的药物,因保留了修饰前活性药物的大部分结构特征,所以在一些传统结构确证手段上具有一定的相似性,同时活性药物与连接臂、连接臂与高分子载体链接的过程中可能产生新的官能团,可通过相应的表征手段进行鉴定。例如修饰前后的药物红外光谱具有类似的活性药物结构的吸收峰,同时与连接臂形成的酯键等新官能团也可以被检测。一般情况下,高分子担载前药均具有一定的空间结构,粒子属性应是关注的重点。高分子前药是否具有自组装成微球的性质,批内粒子均一性,批间粒子一致性等均需进行详细的研究。可采用电镜等分析方法利用统计学手段对粒子属性进行充分的评估。
5.4 高分子前药的特性考察
高分子担载药物除考察常规稳定性项目外,应该在关键时间点重点考察药物特性指标。如粒子属性、药物的释放行为等。由于高分子前药的自组装性能稳定性相对较差,不同时间节点粒子粒径、形貌是否存在差异,进而影响药物的释放行为,需做进一步考察。同时由于高分子载体的特殊性,不同时间节点载体的溶胀行为可能存在一定差异,需进行相应的考察,为临床用药的溶解时间提供相应的依据。
6 结 语
本综述以抗肿瘤药物为例,介绍了几种不同机制的高分子前药,该类药物利用前药和纳米技术的优势,提高了抗肿瘤药物的传递效率。高分子前药涉及到高分子、药学等多学科,通过加强学科间的交流,增强对各种风险点的识别和把控,强化质量源于设计的理念,可有效促进该类药物的研发,为临床用药提供更多的选择性。
