β-内酰胺酶抑制剂的研究进展
2019-08-22匡楠楠张鹤营谢书宇潘源虎袁宗辉
严 勉,匡楠楠,张鹤营,谢书宇,瞿 伟,潘源虎,袁宗辉
(国家兽药残留基准实验室(HZAU),华中农业大学国家兽药安全评价实验室,武汉430070)
β-内酰胺类抗生素在治疗多种临床致病菌感染中发挥着重要作用,然而细菌耐药性的出现使这类药物失去抗菌活性,甚至是耐酶的碳青霉烯类抗菌药也不断出现耐药菌[1-2]。β-内酰胺类抗生素耐药的最主要机制是细菌产生β-内酰胺酶,该酶能与β-内酰胺类抗生素的β-内酰胺环上的羰基共价结合,使其水解、灭活。目前已知的β-内酰胺酶数量有300余种,根据Bush分类法,β-内酰胺酶以优选底物和抑制剂谱的不同分为4组。Ambler根据各自氨基酸同源性序列将其分为A、B、C、D 4类,其中A、C、D类的活性基团为丝氨酸,B类的活性基团为Zn2+(表1)。目前临床上应用的β-内酰胺酶抑制剂如克拉维酸、舒巴坦、他唑巴坦等通过与β-内酰胺酶发生作用,阻断其对β-酰胺环的破坏,有效地对抗了细菌的耐药性,然而β-内酰胺酶的多样化及不利突变使上述三种抑制剂逐渐失去作用。近几年来,基于竞争性抑制剂的设计理念,通过提高抑制剂与酶的亲和力及复合物的稳定性进行活性药物筛选,取得了很大的进展;同时新的酶失活机制报道也为新型抑制剂的设计提供新的思路[3-5]。就近五年来的基于β-内酰胺酶水解机制的β-内酰胺酶抑制剂设计合成及活性研究作一概述,以期为新型化合物的研发提供借鉴。
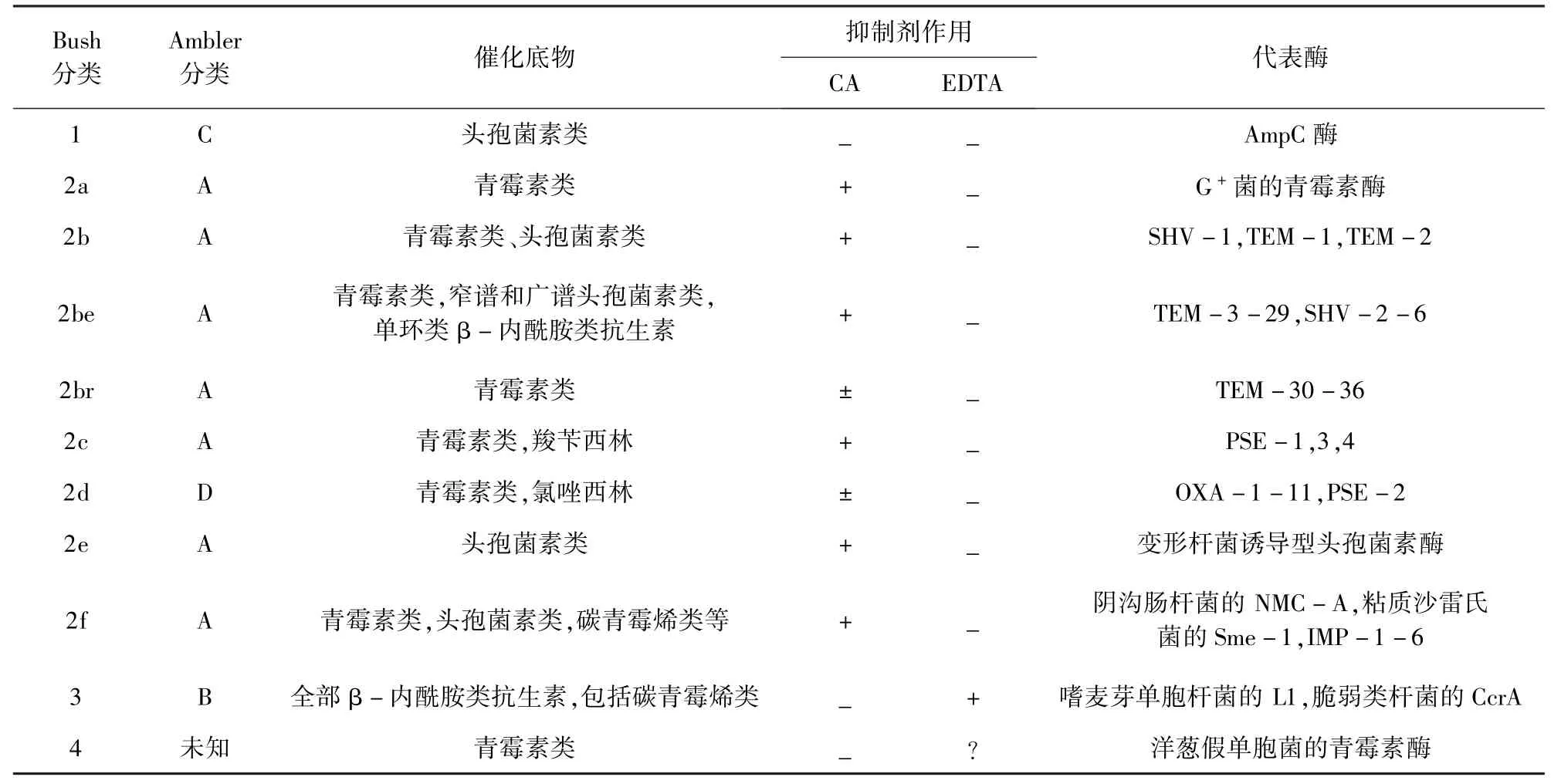
表1 β-内酰胺酶的分类[6-8]Tab 1 Classification of β -lactamase
1 A、C、D类β-内酰胺酶抑制剂
目前上市或在研药物多为A、C、D类β-内酰胺酶抑制剂,按结构可划分为青霉素和头孢菌素类、青霉烯类、单环类、青霉烷砜类及非β-内酰胺环类等。
1.1 青霉素及头孢菌素类似物 该类化合物的构效关系研究多集中于C-3/7位(头孢菌素类)和C-3/6位(青霉素类)。Zhang等合成了一系列硫亚胺头孢菌素类似物,此类化合物的3位取代基在与酶识别过程中起关键作用,构效关系表明以芳香类大基团连接亚胺,可增强对C类头孢菌素酶和TEM-1的抑制活性,其中化合物1和2以2-吡啶取代连接C-7硫亚胺,对野生型枯草芽孢杆菌C类酶的抑制作用优于他唑巴坦,与头孢拉定配伍,对MSSA(methicillin-sensitive Staphylococcus aureus)和肺炎克雷伯菌的MIC值降低2~4倍[9]。Margherita等从6-氨基青霉烷酸合成氮杂环丁酮衍生物3,对沙门氏菌有微弱的抑制作用(MIC50>64 μg/mL),对金黄色葡萄球菌和表皮葡萄球菌的MIC降低2~3 倍 ( 相 比 于 氨 苄 西 林)[10]。 S-649266(Cefiderocol)的C-6位取代与头孢他啶一致,C-3位引入邻苯二酚,进入细菌后可螯合三价铁,使外界铁离子不断进入细胞内。该化合物对多种耐药肠杆菌(含酶 KPC-、VIM-、IMP-)有效,甚至是含NDM-1大肠杆菌、肺炎克雷伯菌[11],且对KPC-3酶较为稳定,相比于美罗培南,200 μM的浓度下水解速率减弱3倍[12](图1)。

图1 青霉素和头孢菌素类衍生物Fig 1 Derivatives of penicillin and cephalosporins
1.2 青霉素砜类 代表药物有舒巴坦和他唑巴坦,它们能与β-内酰胺酶发生非共价结合形成Michaelis复合体,然后70位丝氨酸的羟基进攻抑制剂内酰胺环上的羰基,导致内酰胺环开环形成酰化产物,接着恶唑环开环生成酮衍生物,最后在130位Ser的作用下经过多步重排反应,生成了不可被水解的酶和β-内酰胺类抗生素的多种酰化产物,最终使酶失去活性。戊二酸单甲酯取代物SA2-13,可与SHV-1的某些氨基酸形成盐桥,脱乙酰化速率比他唑巴坦低10倍左右[8]。PSR-3-226的末端羧酸以酰胺取代,能够与KPC-2活性位点构成反式烯胺,可作为靶向KPC-2酶抑制剂[13]。通过分子模拟发现LN-1-255结构中的C-3位基团、C-2位基团及吡啶环与OXA-1稳定结合,疏水性苯环有利于和氨基酸Trp102及Met99的互作,能有效抑制D类酶(OXA-1、-10、-14、-17及碳青霉烯酶OXA-24/40)的活性[14],其对OXA-48抑制活性明显优于他唑巴坦(IC50=3nM)(图2)。
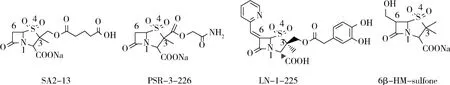
图2 青霉砜类衍生物Fig 2 Penicilla-sulfone derivatives
1.3 青霉烯类(碳青霉烯类、氧青霉烯类) 该类化合物是一种兼具青霉素和头孢菌素骨架结构的的广谱抗菌药,1位S原子以C/O取代,C2-C3引入双键,以增强化合物的稳定性,代表药物有亚胺培南和美罗培南(图3)。此类活性化合物的筛选集中于青霉烯的6位杂环取代、碳青霉烯的3位巯基杂环取代及氧青霉烯的2位取代[8]。Miller等以叔丁基取代C-2位,以不同的烷烯基取代氧青霉烯C-6位筛选活性化合物,C-6位共轭双键是对靶酶特异性识别的关键结构位点(尤其是A类酶)。取代基类型和构像对酶的抑制活性同样重要,其中化合物4和5呈广谱抗菌作用,但光及水解稳定性有待解决[15]。
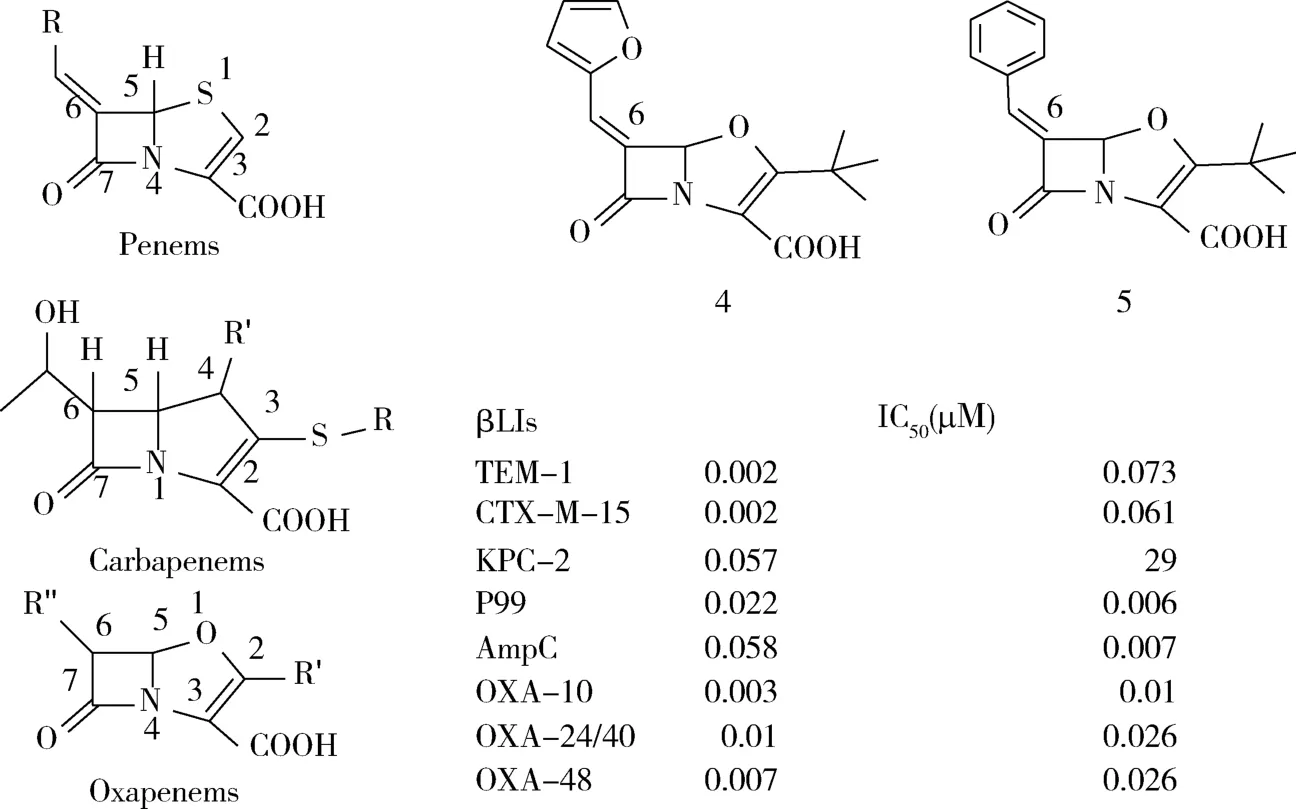
图3 青霉烯类衍生物及其对β-内酰胺酶抑制活性Fig 3 Penicillene derivatives and their inhibitory activity to beta-lactamase
1.4 非β-内酰胺环抑制剂 该类化合物无典型的内酰胺环结构,通过模拟酶与底物(抗菌药)的作用位点,与酶活性位点发生竞争性互作而呈现抗菌作用,包括二氮杂二环辛烷(DBOs)、硼酸类和无特定结构的其他化合物。
1.4.1 二氮杂二环辛烷 (DBOs) 阿维巴坦(Avibactam,AVI,图4)是首个进入临床研究的二氮杂二环辛烷(DBOs)非β-内酰胺环抑制剂,该药与头孢他啶组成的复方制剂已在FDA批准上市。MK-7655作用机制与其相似,结构上以吡啶连接酰胺的NH2,与亚胺培南和西司他汀合用已进入三期临床研究。OP0595是在AVI的2位酰胺NH上连接氨乙氧基,这种烷氨基侧链并不能增强其与酶活性口袋的相互作用,但可能与后者底端存在疏水作用,因此其对A类酶(TEM-、CTX-Ms、KPC-2)的抑制活性与阿维巴坦相近,对C类酶的抑制活性较好(AmpC、CMY-2),与哌拉西林、头孢吡肟、氨曲南呈现协同作用[16]。该化合物单用无活性,和头孢吡肟合用对大肠杆菌CTX-M-15和肺炎克雷伯菌KPC-有效[17],还可抑制肠杆菌科的青霉素结合蛋白PBP2,有学者认为在研究阿维巴坦类似物活性时,这种抑制作用不容忽略[16,18]。
目前,DBO类似物的结构修饰集中在C-2和N-6位(图4),辛烷的的R1分别以N-环丙基甲氧基甲酰胺、卡巴肼取代、得化合物6和7,两者对TEM-1 和 AmpC 的 IC50均小于 0.1 μM,前者对OXA-2 的活性(1μM)优于后者(≥10 μM)[19]。
1.4.2 硼酸类似物 与经典的水解机制不同,硼原子作为亲电体模拟β-内酰胺环的羰基碳,与酶的丝氨酸催化位点形成可逆的四面络合物,这种共价结合物与传统机制中过渡态结构类似(图5)[20]。
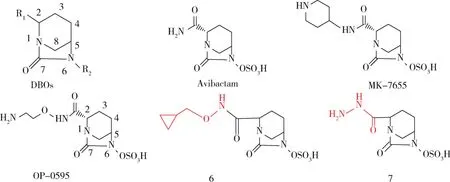
图4 二氮杂二环辛烷(DBOs)类衍生物Fig 4 DBOs derivatives
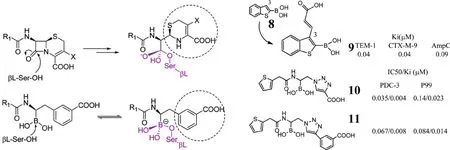
图5 硼酸类衍生物Fig 5 Boric acid derivatives
化合物8的3位引入极性羧酸侧链,可与TEM-1(Arg244和 Ser235),AmpC(Asn346 和 Asn289)及CTX-M-9(Arg276)关键氨基酸作用,增强结合亲和力,扩大广谱作用谱。基于此项研究,Donatella等合成了化合物9,其对TEM-1和CTX-M-9的活性可增强1250倍,但稳定性有待解决[21]。Caselli等以三唑环连接C-2,对C类酶呈现较好的亲和力和抑制活性,其中化合物10和11活性最好,化合物10与三种 C类酶(AmpC、PDC-3和P99)的互作氨基酸高度保守,以 4 μg/mL 的浓度可恢复头孢噻肟对大肠杆菌的活性(PDC-3和P99)[20]。
研究发现硼酸类化合物与TEM-1复合物中存在环化硼酸酯结构,Hecker等设计合成了一系列硼酸酯类化合物,分别以脂肪族、芳香族及氮杂环取代R基团(图6)。 RTX7009(vaborbactam)以噻吩连接氨基(图6),对A和C酶的Ki值可达nM级。该化合物单用无活性,与头孢吡肟合用可对多种耐药菌有效,且无明显毒性[22],与比阿培南配伍对产KPC酶的耐药肠杆菌有效[23]。另一类硼酸酯类化合物12结构中引入杂环,与AmpC增加氢键互作,苯环上双甲基与OXA-10疏水氨基酸互补,恢复头孢他啶对含酶的大肠杆菌和枸橼酸杆菌敏感性[24]。
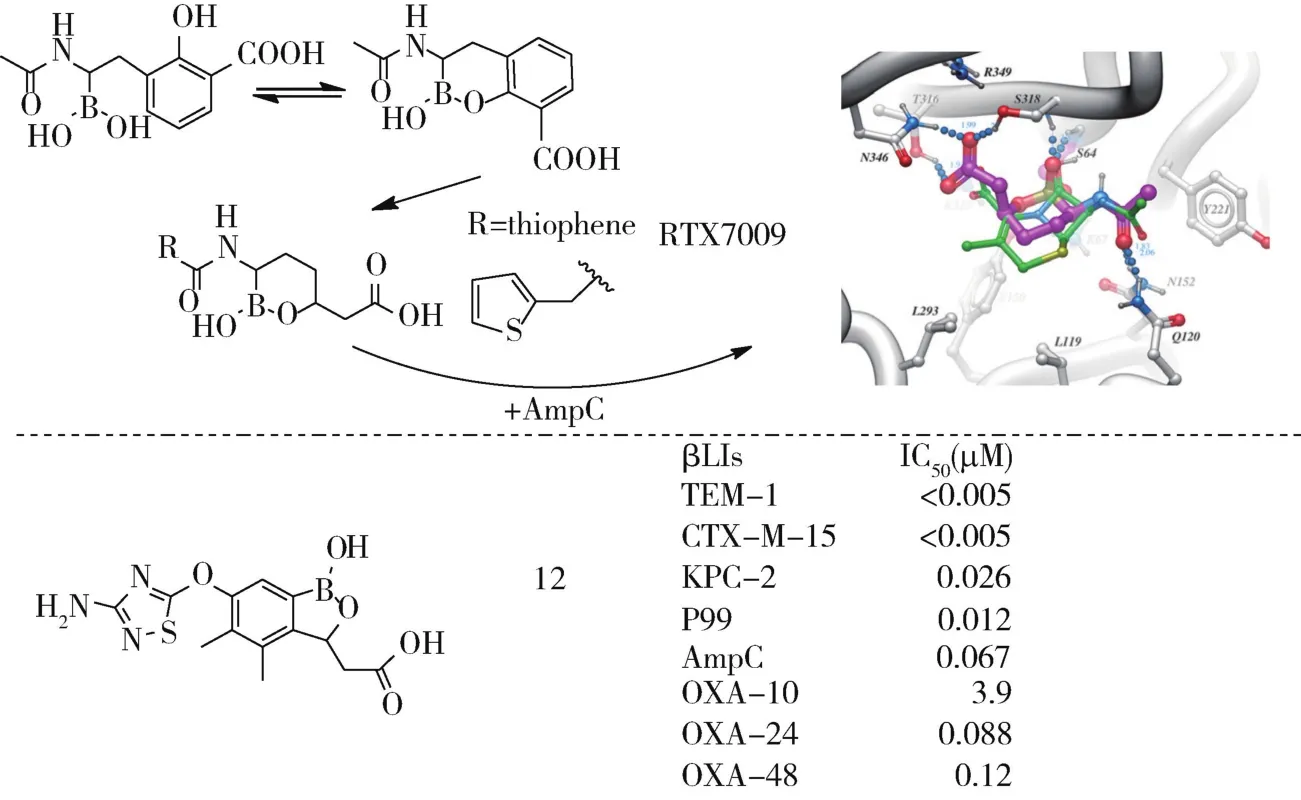
图6 硼酸酯类衍生物Fig 6 Borate ester derivatives
1.5 其他A、C、D类β-内酰胺酶抑制剂 O-芳基羧酸酯异羟肟酸是针对靶向DD-肽酶活性位点而设计的化合物,因C类β-内酰胺酶与后者的活性位点关键氨基酸相似,Tilvawala等合成了羟胺衍生物13(图7),该化合物与P99酶发生快速乙酰化的过程中离去苯氧基团而保留酰基羟胺与C类酶的Ser64共价结合,其侧链极性基团羧酸和氨基与活性位点附近 Ω loop(Gly320、Ser212和Arg204)发生有利互作形成稳定的过渡中间态使其脱乙酰过程减慢。酰化酶与丝氨酸的Ω loop间的这种互作结合可为新型抑制剂设计提供新思路[25]。Tan等合成了噻吩肟磷酸酯衍生物14和15,其磷酸基团与丝氨酸酶形成共价复合物,对 C酶(P99和AmpC)的活性最佳,与亚胺培南合用对产AmpC的绿脓杆菌呈剂量依赖型作用,对A和D酶的作用相对较弱[26]。

图7 其他丝氨酸酶抑制剂Fig 7 Other serinase inhibitors
四唑类化合物是基于CTX-M内酰胺酶两个研究热点—Pro167的疏水作用和Asp240极性末端设计一种新的非共价结合的化合物,其作用与传统β-内酰胺类抗生素的C-(3)4'位羧酸一致,化合物16(图7)在苯环上引入疏水基团 CF3增加对Pro167的亲和力,苯并咪唑作为氢键供体与Asp240互作,使其对大肠杆菌CTX-M-9的亲和力显著提高(Ki=89 nM),与头孢噻肟联用恢复其对(CTX-M-9)的敏感性,MIC 降低64 倍[27]。
2 B类β-内酰胺酶抑制剂
B类β-内酰胺酶抑制剂多为锌离子结合剂,其选择性和耐受性问题在于Zn2+螯合作用和酶活性位点氨基酸与其他含锌蛋白酶的差异性,因此活性研究需区分金属离子螯合作用和配体-酶复合物的形成作用[28]。
2.1 硫醇类化合物 该类化合物主要通过硫醇络合Zn2+发挥作用。Arjomandi等将氨基酸(Phe、Trp、Tyr)与硫醇化合物连接合成一系列衍生物,根据酶动力学结果筛选出化合物17,与亚胺培南合用对含IMP-1大肠杆菌、含IMP-4的阴沟肠杆菌的MIC 值分别降低5.2、3.9 倍(0.38 μg/mL),且以短链连接时活性较好(Ki=0.086 μM)[29]。 Liu 等将氨基酸连接于噻吩和巯基乙酸之间合成硫酯化合物18,可选择性作用于L1酶,其中化合物19和20与L1的作用方式相同[30],而以4-联苯取代噻吩环,抑制活性更好[31](图 8)。
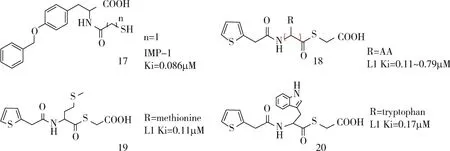
图8 硫醇类衍生物Fig 8 Mercaptan derivatives
2.2 三唑硫代乙酰胺 Zhang等设计了18种硫代乙酰胺类似物,分别对骨架21(图9)的R1和R2进行修饰,所有化合物对嗜麦芽窄食单胞菌金属酶L1(Ki<2 μm)有效,部分作为广谱抑制剂对四种受试酶 CcrA、NDM-1、ImiS、L1皆有效,其中化合物22优先通过三唑基团定位到Zn2+,2位羟基与活性位点氨基酸残基 Lys224(CcrA、NDM-1和ImiS)或Ser221(L1)结合(广谱抗菌的原因),对四种酶的 Ki值可达到 0.30 ±0.09、0.43 ±0.03、2.2 ±0.2、0.073 ±0.007 μM[32],R1 以芳香族羧酸取代可提高对ImiS的活性,如化合物23,Ki值为1.2 μM[33];R2 芳香环4 位取代物活性优于 2 位取代,如化合物24,左侧芳香环上氨基则与NDM-1的 Lys 211 互作,Ki为0.49 μM[34](图9)。
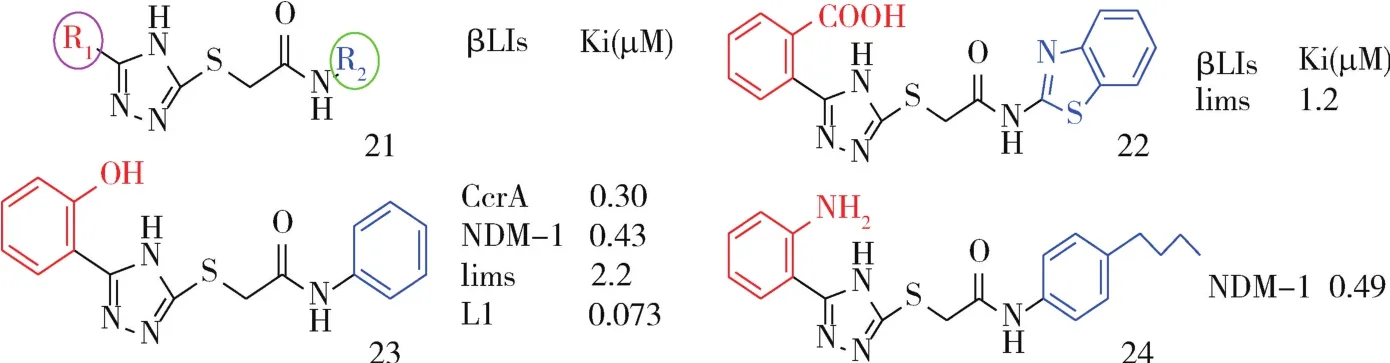
图9 三唑硫代乙酰胺衍生物Fig 9 Triazole thioacetamide derivatives
2.3 金属络合剂 这类化合物通过络合金属酶活性位点Zn2+发挥抑制作用,主要包括羧酸类和氮杂环化合物(图10)。EDTA可抑制NDM-1的活性(IC50=1.6 μM)[35],但存在毒性作用。 近年来研究的新化合物 NOTA、DOTA、NODAGA、ME1071等(图 10)能与碳青霉烯类合用,起到增敏作用[36-38]。其中NODAGA的1位末端羧基分别以四肽和八肽连接,增效作用最佳,化合物TPEN对Zn2+的亲和作用优于DPA,但细胞的氧化应激毒性作用存在弊端[36]。咪唑衍生物5-甲基-2-苯氧甲-3-H-咪唑-4-羧酸(PIMA)结构与青霉素V相似,可螯合锌和铁离子[39]。目前,大环羧酸类对锌离子的亲和性及安全性较好,临床开发价值较高,但对靶酶选择性问题尚待解决。
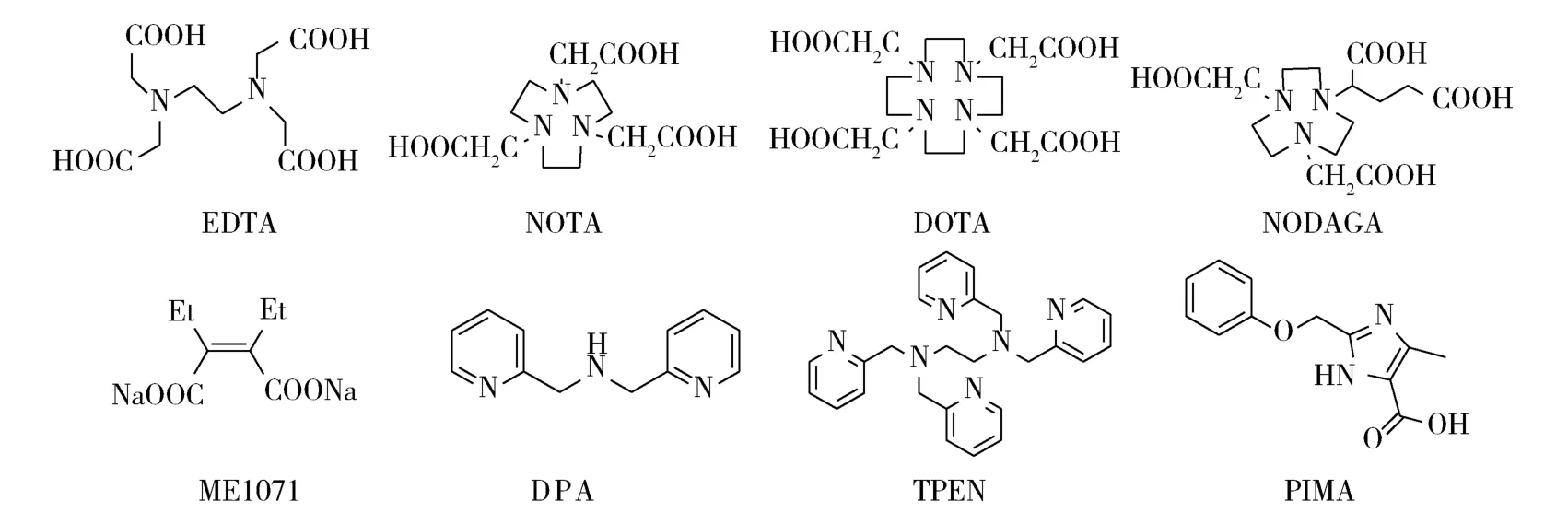
图10 金属络合剂Fig 10 Metal complexing agent
2.4 多肽类 Caitlyn等合成了一系列含精氨酸的肽类,可作为选择性抑制剂对酶-底物复合物具有较高的亲和力,通过引发酶的聚合反应抑制其活性。该抑制剂活性主要受精氨酸数量及N端氨基酸类型的影响,对VIM-2的抑制活性可达到nM级,明显优于IMP-1[40]。Xiao等基于金属酶 VIM-2活性位点两个loop环(Phe42-Ala45和Ile180-Trp199)的定位和识别作用,从组合肽数据库筛选出三肽甲硫酰胺-半胱酰胺-色氨酸(25)和酪酰胺-半胱酰胺-色氨酸(26),IC50分别为18.15 μM 和 52.9 μM)(图 11),分子对接中半胱氨酸巯基对金属的螯合作用不同导致其抑制活性存在差异性,前者甲硫氨酸氨基与Asp193形成氢键,使loop环更加稳定,抑制活性更好[41]。
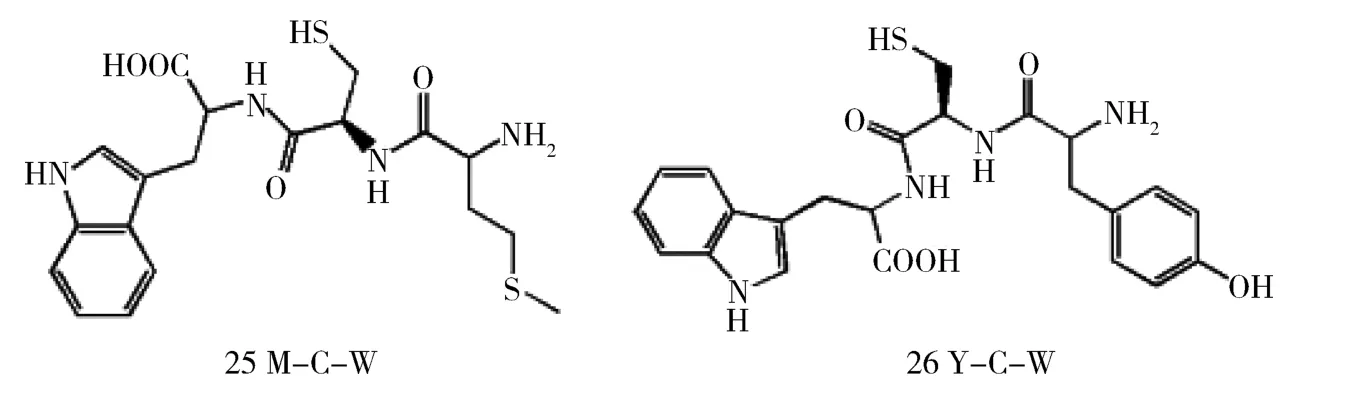
图11 三肽类化合物化学结构Fig 11 Chemical structure of three peptide compounds
2.5 其他金属酶抑制剂 Christopeit等首次报道对NDM-1有效且可逆的共价非β-内酰胺环化合物27,Ki值为 76 μM (图 12),其醛基与 Lys224末端氨基形成共价复合物,色酮羰基氧与Zn2+互作,同时与Trp87、Ile35和Val67存在疏水作用力。这种作用机制为β-内酰胺酶抑制剂的设计提供新的思路,但醛基相对不稳定,故有待优化。Mark等合成了一系列N-甲巯基衍生物,不含羧基或磺酸基等可电离基团,对NDM-1和VIM-1的Ki值在25~0.03 μM,多数化合物对结核杆菌有效,其中化合物28和29对多种酶呈现抑制作用[42]。Yang等设计合成化合物β-内磷酸环衍生物化合物28,对IMP-1,CcrA,L1,NDM-1 和Bla2 有抑制作用。化合物30磷酸中一个氧原子结合Zn1,N1结合Zn2,C-7位羧酸与金属酶的Lys224侧链形成盐桥(L1除外)。
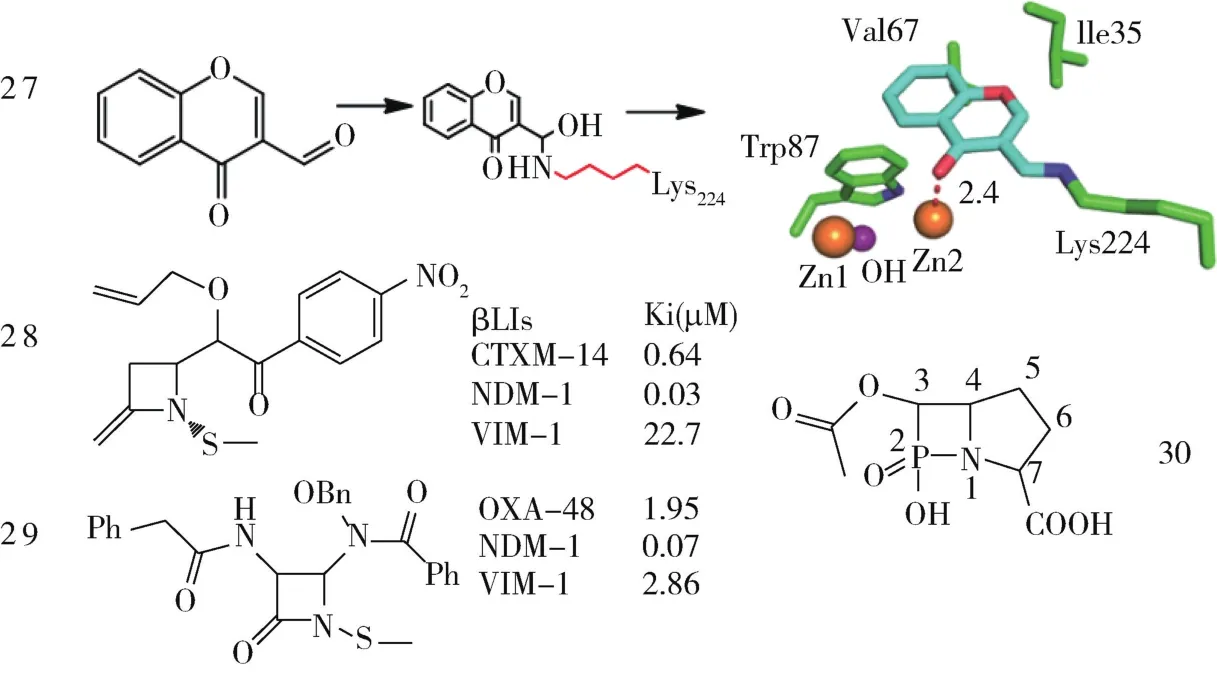
图12 其他金属酶抑制剂Fig 12 Other metalloenzyme inhibitors
3 展 望
β-内酰胺类抗生素在抗感染领域发挥着重要的作用,而耐药菌的增多使该类抗生素在临床上的应用大打折扣,所以新型β-内酰胺酶抑制剂不断开发和应用已是抵抗该类细菌耐药性的主要方法。目前,新型β-内酰胺酶抑制剂研究无论是基于片段设计,还是对已有化合物的结构修饰研究,都多通过与靶酶的酶抑动力学及抑制活性评价初步筛选候选物,再以共晶和体外抗菌活性测定,结合细胞毒性数据,对化合物进行综合评定。近年来,基于酶关键作用氨基酸的活性化合物筛选取得了一定进展,如金属酶VIM-2的两个loop环及CTXMs酶的Pro167和Asp240等,能够与这些氨基酸形成稳定的中间态是活性化合物筛选重要条件。由于金属酶多具有广谱水解作用,空间结构及水解机制相似,对相同活性位点氨基酸展开针对性的互作研究有助于广谱抑制剂的筛选,如Lys224、Cys221等。虽然目前临床上未见对四种β-内酰胺酶有效的化合物,但依据各类化合物对靶酶的选择性来进行合理的配伍,不失为一种理想的应对策略,有研究表明阿维巴坦/头孢他唑与氨曲南合用对MβL耐药肠杆菌有效,甚至是含NDM-1和其他类酶的耐药菌。耐药突变及耐药基因的传播也是当然无法完全避免的关键问题,因此开发出对野生型及突变体同样有效的抑制剂,从源头上减缓耐药性的产生也是今后研究的一大趋势。
