心肌梗死后的心功能改善:有氧运动干预发挥效应新视角
2019-08-21王友华马美田振军
王友华 马美 田振军
陕西师范大学体育学院(西安710119)
近些年来,随着生活水平的提高,肥胖、高脂血症、糖尿病日益增加,它们是诱发心血管疾病(cardiovascular disease,CVD)的共同土壤。心血管疾病如心肌梗死(myocardial infarction,MI)及其并发的心律失常和心力衰竭等已成为威胁人类健康的头号杀手,一直以来都是国内外学者研究的重点课题,但是如何治疗和预防心肌梗死及其并发症,依旧是国际众多学者和科研工作者亟待解决的科学难题[1-3]。研究表明,运动训练能改善心功能,临床上已把运动作为治疗与预防心肌梗死的重要干预手段,但其机制仍在探索中。本研究团队十多年来一直致力于运动改善心血管功能的生物学机制研究,证实运动改善心血管功能与心脏神经系统调控和血管功能改善密切相关[4,5]。本文在我们前期研究的基础上,通过综述心肌梗死后交感神经与副交感神经重构、线粒体自噬变化与心肌梗死心脏功能恶化的相关性的研究报道,进一步论述有氧运动干预对心脏功能改善的生物学机制,为临床实践中心肌梗死患者的治疗提供理论依据和新的视角。
1 有氧运动对心肌梗死心脏神经系统调控的生物学机制
1.1 交感神经调控与心脏功能变化
运动心脏神经生物学领域的研究一直是心血管研究中的热点课题。心脏受自主神经支配,包括交感神经和副交感神经调节心脏传导、节律和收缩性。交感神经和副交感神经失衡往往是导致心律失常和心力衰竭的重要因素。心脏神经系统的形态、结构和受体蛋白表达的变化与心脏功能障碍和猝死密切相关。
MI后心脏自主神经的变化包括交感神经和副交感神经的重构,其中交感神经再生占主导地位;交感神经活性增加而副交感神经活性降低,最终造成心脏自主神经支配的不均衡分布。心肌梗死后交感神经重构表现为神经生长和交感神经过度支配。研究发现,炎症反应是MI后发生交感神经重构的重要因素,心梗后的心肌坏死可导致炎症反应,大量的内皮素-1(endothelin 1,ET-1)和肿瘤坏死因子-α(tumor necrosis factorα,TNF-α)等炎症细胞因子在心脏中表达[6]。MI 后的炎性细胞促进神经生长因子(nerve growth factor,NGF)合成,其机制是炎性细胞因子激活核转录因子kB(nuclear factor-kappa B,NF-kB)调节炎症反应和NGF信号通路[7]。NGF 的合成和释放可以引发心肌梗死交感神经重构。NGF 通过交感神经元中的p75神经营养因子受体和TrkA受体起作用,刺激轴突生长。信号转导和转录激活因子3(signal transducer and activator of transcription,STAT3)是心肌梗死后心脏中NGF诱导的交感神经再生所必需的[6]。
室性心律失常是心肌梗死后心脏性猝死的常见原因,交感神经重构会引发室性心律失常的发生[6]。因此,抑制交感神经可以改善心肌梗死后交感神经的重构,并且降低疾病及猝死的发生率。目前,干预交感神经重塑及其机制的研究已经成为热点,为MI后室性心律失常的治疗和预防提供了一个新的方向。大量研究表明,有氧运动可以降低心脏交感神经张力,减少室性心律失常的发生[8,9]。本研究团队前期研究发现,MI导致心功能显著降低,有氧运动通过活化MI后内皮型一氧化氮合酶(endothelial nitric oxide synthase,NOS2)和神经型一氧化氮合酶(neuronal nitric oxide synthase,NOS1)来恢复β3-肾上腺素能受体/β1-肾上腺素能受体(β3-adrenergic receptor/β1-adrenergic receptor,β3-AR/β1-AR)平衡并增加β3-AR表达,从而抑制心肌交感神经重构,有效改善MI 大鼠心脏功能[4]。炎症反应是MI后交感神经重构的一个关键方面,抗炎可以减轻MI 后交感神经重构[10]。有文献报道,有氧运动可减少心衰患者血液中的炎症因子的表达,表明有氧运动对心衰患者心脏的保护作用与其抗炎作用紧密相关[11]。新近文献报道,运动干预后转基因镰状细胞小鼠中白介素-1β(interleukin-1β,IL-1β)和ET-1 mRNA 表达减少,表明运动可以减轻这种全身性炎症疾病状态,运动具有抗炎作用[12]。另外,有氧运动减少了MI 大鼠左心室和比目鱼肌中的干扰素-γ(interferon γ,IFN-γ)、白介素-6(interleukin-6,IL-6)、IL-1β和TNF-α,证实了有氧运动抑制炎症反应保护心脏和肌肉[13]。心肌梗死后有氧运动通过下调梗死灶周围组织NGF 的表达和抑制核转录因子及其转录系统的激活,减少了MI后心肌炎症反应及其连锁效应,最终抑制MI后交感神经重构改善心脏功能。
综上,MI 后交感神经的重构是导致心脏交感神经过度兴奋、恶化心肌梗死的重要因素。有氧运动通过抑制交感神经的重构削弱交感神经过度增生,达到改善心脏功能的效果。
1.2 副交感神经调控与心脏功能变化
心脏副交感神经对心脏的调节起重要作用,心脏副交感神经支配心脏的能力降低将使自主神经失衡,是诱导心血管疾病恶化的重要因素。目前,有关心脏副交感神经的研究主要是在心肌缺血后特定时间的神经形态及其分布的改变,而对MI后副交感神经重构及运动干预对其的影响还需进一步研究。
有文献报道,MI 后副交感神经的形态和分布发生了与交感神经相似的变化[14]。在猪的心梗模型中发现,MI后发生了副交感神经失支配的现象。大鼠急性心肌梗死可以引起交感神经激活过度,降低了副交感神经的张力,观察胆碱能神经分布后发现,心梗后右室副交感神经纤维密度逐渐下降,与MI早期易发生心源性猝死的时间相对应[14],说明自主神经失衡与心源性猝死关系密切。另有研究发现,心肌梗死后交感神经过度激活,副交感神经活性降低,则心率变异性(heart rate variability,HRV)下降(自主神经失衡),发生室性心律失常[15]。大量文献表明,心梗后张力反射敏感性和心率变异性降低越大的患者,恶性心律失常和猝死的发生率就会明显增加。
有学者发现,MI 后有氧运动可以提高心脏副交感神经的兴奋性同时降低交感神经兴奋水平,提示有氧运动对交感神经的抑制作用及对副交感神经的活化效应。老年心肌梗死患者早期进行运动训练后,运动贮量明显提高,自主神经调节和心率变异性改善,心肌梗死猝死率及病死率降低,患者无明显不良反应,生活质量提高[16]。本研究团队先前已经证实,有氧训练可提高大鼠心脏功能,上调胆碱能神经和M2毒蕈碱乙酰胆碱受体(M2muscarinic acetylcholine receptor,M2receptor),推测有氧运动造成心脏功能的改变与运动诱导心脏副交感神经调控能力改变有关[17]。而且,有氧运动可以逆转高脂血症导致的心肌胆碱能神经和M2受体降低,活化副交感神经,改善心脏功能[18]。并且有氧运动可以上调心梗大鼠内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)和下调心梗大鼠TNFα的表达,提高心脏负性肌力和降低炎症反应[5,19]。在此基础上,最新研究发现有氧运动上调心肌梗死大鼠副交感神经M2受体和α7烟碱乙酰胆碱受体(α 7 nicotinic acetylcholine receptor,α7nAChR),提高副交感神经支配能力,进而改善心脏功能(此数据尚未发表)。有氧运动激活α7nAChR 引发胆碱能抗炎途径,导致心梗大鼠TNF-α的表达降低。据报道α7nAChR 在内皮细胞的增殖中上调,并且α7nAChR 敲除的小鼠血管生成减弱[20]。推断运动通过上调α7nAChR 促进血管生成,从而恢复压力反射功能,尤其是迷走神经活动,进而改善心脏功能。运动可以上调心脏中的神经型一氧化氮合酶(neuronal nitric oxide synthase,nNOS)和eNOS,从而引起大鼠心脏中M2受体基因的表达增加,此机制可能是运动提高副交感神经活性的重要因素[21,22]。有氧运动通过提高大鼠副交感神经张力和增强副交感神经抗炎效应来改善MI 心功能。有氧运动作用机制还有待进一步深入研究。
心肌梗死的恶化常伴随着心脏副交感神经支配的降低或失活,有氧运动作为有效提高心脏副交感神经的干预措施,对于心肌梗死患者的康复意义重大。有氧运动对心血管疾病患者副交感神经调节影响的深入研究将为从副交感神经的角度预防和治疗心肌梗死提供新的思路和靶点。
2 有氧运动对心肌梗死心脏线粒体自噬调控的生物学机制
线粒体质量控制系统对于维持线粒体功能至关重要。在细胞器水平上,包括线粒体生物合成,融合和裂变,自噬。细胞通过自噬机制选择性地降解线粒体的途径称为线粒体自噬。
正常情况下,心肌细胞的自噬水平很低。在动物心肌梗死模型中,自噬增强,自噬体标记物微管相关蛋白1 轻链3Ⅱ(microtubule-associated protein 1 light chain 3Ⅱ,LC3Ⅱ)、P62 和组织蛋白酶D 表达均上调;自噬在心肌梗死1周后显著激活,在心肌梗死后3周形成自噬体[23]。研究显示,在心肌梗死过程中,自噬的调控主要是通过腺苷酸活化蛋白激酶(AMP-activated proteinkinase,AMPK)来激活并控制的,AMPK 被磷酸化后激活自噬通路,致使自噬反应增强,进而促进心肌细胞的存活[24]。另有研究发现,在心梗早期阶段,AMPK/雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路激活,自噬上调,从而维持心肌细胞的稳态,改善心脏功能[25]。心肌梗死可以诱发氧化应激,从而导致活性氧(reactive oxygen species,ROS)的大量产生,进而引起自噬;ROS可以介导自噬生成过程中的各个信号通路,是自噬的主要诱导者[26-28]。心肌梗死再灌注阶段通过Ⅲ型磷脂酰肌醇3激酶(phosphatidylinositol3-kinase class Ⅲ,ClassⅢPI3K)/Beclin1 途径进一步激活自噬,Beclin1 过度激活,心肌细胞自噬大量上调,这会抑制自噬小体和溶酶体的融合,所以形成的自噬体增多并不能被及时清除而聚集在胞质内,自噬流被明显抑制,最终启动凋亡通路,造成细胞死亡,引起心脏收缩功能障碍最终导致心力衰竭[25]。虽然自噬标记物Beclin1的表达增多会过度激活自噬,对心肌细胞起到损伤作用,但具体的信号通路需要继续研究证实。新近研究得出,心肌梗死导致大鼠心力衰竭,自噬流和线粒体功能受损[29]。另有文献报道,大鼠心肌梗死后LC3II/微管相关蛋白1轻链3I(microtubule-associated protein 1 light chain 3I,LC3I)的水平升高,心肌线粒体和自噬小体的数目增加,Parkin活性的降低导致线粒体自噬流障碍以及正常形态破坏[30]。MI后衰老心脏的线粒体自噬信号相关蛋白LC3II 和P62 上调以及假定激酶1(PTEN induced putative kinase 1,PINK1)/Parkin表达抑制。PINK1/Parkin的低表达表明LC3II和P62蛋白水平的积累来自LC3Ⅱ转换的抑制,暗示自噬体降解阻断[31]。在心梗后不同的阶段,自噬会发挥不同的效应。心梗前期,激活的自噬对心肌是有利的,而自噬后期,过度激活的自噬会导致细胞死亡。所以自噬在心肌梗死中所起的作用存在很大争议。自噬是将来预防和治疗心脏疾病的潜在途径,寻找有效的手段调控线粒体自噬,可能是防治心肌梗死的新策略。
有氧运动介导心肌梗死后的过度激活的自噬,对于预防和治疗心血管疾病有重要参考价值。有文献报道,有氧运动显著降低家兔MI组中的LC3Ⅱ表达,并且导致MI 组中LC3Ⅱ与LC3Ⅰ的比例较低,但并未改变假手术组的比例,说明运动改善了MI 兔的心脏功能,促进心肌梗死后堆积的自噬体的降解[32]。又有数据显示,短时间运动可以调节老年心梗小鼠线粒体自噬标志物,包括激活PINK1/Parkin 表达以及降低LC3Ⅱ和P62 水平,PINK1 将Parkin 募集到线粒体,引发线粒体自噬[31]。最近发现小鼠在心肌梗死后进行运动训练,能改善心脏自噬流,之后线粒体数量减少;运动调节线粒体裂变融合平衡和自噬流来改善线粒体质量控制,从而改善MI 小鼠心脏功能[29]。有数据表明,长期运动训练可以减少小鼠心肌梗塞面积并减轻心梗急性期心肌细胞凋亡和自噬,发挥心脏保护作用。此外,运动训练改善心肌葡萄糖和脂质代谢,并进一步增强心梗小鼠的线粒体生物合成,伴随着过氧化物酶体增殖物激活受体γ辅助激活因子-1α(peroxisome proliferator-activated receptor γ coactivator 1α,PGC-1α)的激活[33]。进一步了解运动对心梗心脏保护作用的分子机制可能对于开发新的药物策略挽救心梗急性期的心脏功能具有重要作用。
大量研究表明,有氧运动在心梗后可起到保护心肌的作用,尽管其中机制还有待继续研究[34-38]。有氧运动改善心肌梗死后线粒体自噬,其机制是否与AMPK/mTOR 通路、Beclin1 和classIIIPI3k/Beclin1 途径相关目前还未有文献报道。最新研究发现,FK506结合蛋白8(FKBP8)和微管相关蛋白1 轻链3A(microtubule-associated protein 1 light chain 3A,LC3A)的异位共表达导致以Parkin 非依赖性方式的线粒体自噬[39]。而有氧运动是否通过上调FKBP8蛋白的表达从而促进心肌梗死受损线粒体自噬增加还未见文献报道,这需要后续进一步研究,可能为心梗心脏的防治提供新的靶点。
3 副交感神经刺激(有氧运动)调节线粒体自噬改善心梗心脏功能
有文献报道,副交感神经刺激可以诱导心肌缺血中的M3 毒蕈碱乙酰胆碱受体(M3 muscarinic acetylcholine receptor,M3R)/CaMKKβ激酶/AMPK 途径改善线粒体动力学和功能,表明刺激副交感神经可以上调其受体改善线粒体功能[40]。另外,副交感神经刺激上调乙酰胆碱(acetylcholine ,ACh)活性,促进缺血心肌线粒体生物合成及功能,表明副交感神经刺激上调的ACh 与缺血心肌线粒体功能改善关系密切[41]。副交感神经递质ACh 通过毒蕈碱乙酰胆碱受体(Muscarinic acetylcholine receptor,MAChR)激 活AMPK/mTOR 通路,上调低氧/复氧后自噬数量和功能[42]。还有研究显示,ACh 通过M2受体和PINK1/Parkin 信号通路诱导缺氧/复氧损伤的细胞中的线粒体自噬增加,清除受损线粒体[43]。PINK1是一种线粒体蛋白,在线粒体去极化后被募集并保留在线粒体外膜上,导致Parkin 转位到细胞器。一旦进入线粒体,Parkin 泛素化外膜蛋白,导致自噬蛋白的募集,随后线粒体自噬。PINK1 介导的泛素磷酸化可激活Parkin,从而扩增这一过程[44]。α 7nAChR 的激活,促进心肌缺血大鼠中JAK2/Bcl-2 和PI3K/Bcl-2 级联改善Beclin1 相关的自噬功能障碍,从而减轻心肌缺血中的心脏损伤[45]。心梗降低副交感神经活性,而有氧运动可以激活副交感神经,推测有氧运动通过上调ACh和副交感神经受体及其信号通路促进心梗心脏线粒体自噬,改善心脏功能(图1)。线粒体自噬是改善心肌梗死的不可替代的靶点,有氧运动刺激副交感神经神经活性对心梗心脏线粒体自噬的调控作用还需要大量的研究。
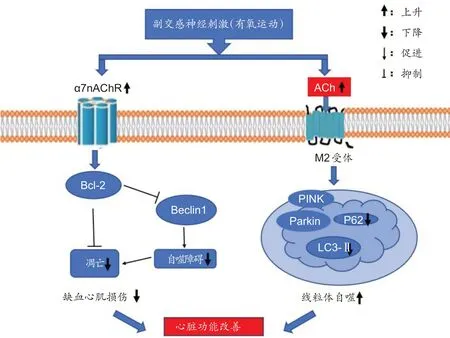
图1 副交感神经刺激(有氧运动)调节缺血心肌自噬改善心脏功能
运动介导的副交感神经改变对线粒体自噬的调节是一个复杂的过程,进一步探究运动介导副交感神经调控线粒体的具体机制和中心环节,将为心肌梗死心脏损伤的临床预防和治疗提供新的思路,具有重大临床价值和现实意义。
4 研究展望
综上所述,心肌梗死后神经、线粒体在恶性室性心律失常和猝死的发生中起重要作用。运动保护梗死后心脏的机制与降低交感神经兴奋性和提高副交感神经兴奋性、调节线粒体自噬水平密切相关(图2)。除此之外,有氧运动对心梗心脏的保护作用还与减轻内质网应激、舒张心脏血管及促进梗死区微血管再生等有关。本团队长期系统从事运动对心肌梗死保护的分子机制研究,获得一些重要成果,如有氧运动改善肠系膜动脉收缩/舒张功能,降低心肌梗死大鼠胰岛素抵抗[46],运动训练激活神经调节蛋白1(NRG1/ErbB)信号促进心梗大鼠模型中的心脏修复等[47]。
近年来,探讨心磷脂酰基转移酶1(Acyl-CoA:lysocardiolipinacyltransferase-1,ALCAT1)与线粒体自噬和代谢改变的研究已经成为热点[48]。有氧运动是否通过调节心肌梗死心脏ALCAT1进而调节线粒体自噬和代谢水平,对于心肌梗死的预防和治疗来说是一个崭新的研究领域。关于有氧运动抑制ALCAT1改善心梗心脏的分子机制以及提高心脏副交感神经功能的具体机制的深入研究将对筛选治疗心肌梗死的有效靶点提供理论依据。有文献报道,内质网应激(endoplasmic reticulum stress,ERS)的增加可以导致线粒体自噬和代谢发生紊乱[49]。内质网(endoplasmic reticulum,ER)在脂质代谢中起调节作用[50]。因此,内质网应激参与脂质代谢和心血管病变的病理生理反应。这提示,抑制ERS可能改善脂代谢紊乱并降低心血管疾病风险。而有氧运动对内质网应激具有很好的抑制作用,但具体机制不详。心肌缺血后ERS 有三条凋亡途径:CHOP、caspase-12及JNK激活,有氧运动改善心脏功能是否与运动抑制ERS 的三条凋亡途径激活有关,还未见文献报道,此方面的研究有待进一步深入。ALCAT1可以导致线粒体损伤,进而引起内质网应激,有氧运动抑制ALCAT1 进而调节线粒体自噬和内质网应激阻止心肌梗死的恶化,这可能是预防治疗心血管疾病的一个关键突破口。
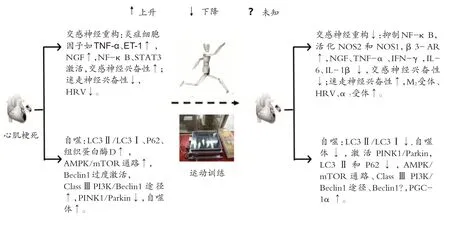
图2 心肌梗死及有氧运动干预后心脏神经重构、线粒体自噬相关蛋白和信号分子的变化
