茶树SRO基因家族的鉴定及表达分析
2019-08-21郭永春王鹏杰陈笛郑玉成陈雪津叶乃兴
郭永春,王鹏杰,陈笛,郑玉成,陈雪津,叶乃兴
茶树基因家族的鉴定及表达分析
郭永春,王鹏杰,陈笛,郑玉成,陈雪津,叶乃兴*
福建农林大学园艺学院/茶学福建省高校重点实验室,福建 福州 350002
(Similar to rcd one)是植物特有的基因家族。本研究利用生物信息学方法从茶树基因组中鉴定获得9个茶树基因家族成员,分别命名为和。9个茶树基因的编码蛋白均具有特征结构域PARP和RST,具有相似的保守基序。系统进化树分析聚分为3组,Ι组包含CsRCD1—4,Ⅱ组包含CsSRO1和CsSRO2,Ⅲ组包含CsSRO3—5。基因结构分析表明每个基因含有4至9个外显子。8个茶树组织转录组数据分析表明,和可能在茶树不同发育阶段具有重要作用;大多数基因在根和成熟叶中较高表达。上游启动子区域分析发现大量与植物发育、激素及胁迫响应密切相关的顺式作用元件,进一步对基因在干旱和脱落酸处理下的表达模式进行分析发现,9个基因均被诱导表达,基因可能与茶树抗旱密切相关。
茶树;;系统进化;干旱胁迫
植物在遭受逆境胁迫时,能够诱导逆境响应基因表达,以在复杂多变的环境生存[1],在这些过程中,通过应激反应参与多条调控网络发挥作用[2-3]。SROs通常包含高度保守的PARP[poly(ADP ribose)polymerase catalytic;PS51059]催化中心和C末端的RST(RCD1-SRO-TAF4;PF12174)保守结构域[4],部分SRO还含有N末端的WWE(PS50918)结构域[5]。
在拟南芥中,家族有6个成员,分别为和[6]。是第一个被鉴定出的拟南芥SRO家族成员[7];可通过与细胞核中的转录因子相互作用,参与植物脱落酸(Abscisic acid,ABA)信号通路介导的干旱响应,还可以通过参与ABA、乙烯(Ethylene,ETH)、茉莉酸甲酯(Methyl Jasmonate,MEJA)等激素信号通路,调控植物发育[8-9]。和,这两个同源基因在不同胁迫条件下的功能存在部分冗余[10],参与非生物胁迫响应,其突变体对渗透和氧化等胁迫具有较强的抵抗能力[11-12];与转录因子相互作用调控基因表达,过表达可以通过降低根中H2O2的水平以增加转基因植株的耐盐性[13];可以响应强光、盐处理及臭氧胁迫[4];没有明确的功能报道[14]。SROs在苹果、水稻、小麦、玉米、陆地棉、番茄等作物中也有部分研究,例如,在苹果中,可通过ABA信号通路调节气孔孔径,耐受干旱胁迫,并且调节根系生长[15];在水稻中,通过调控SNAC1和DST促进水稻气孔关闭和H2O2积累,参与干旱和氧化应激反应[16];在小麦中,可以通过调节植物体内的氧化还原平衡来提高其耐旱能力[2]。越来越多的植物基因家族得到鉴定,SROs在干旱胁迫响应下的作用机制也日益清楚。
尽管前人对各种植物基因已进行大量研究,但有关茶树的基因研究仍是空白。茶树[(L.) O. Kuntze]是一种多年生常绿木本植物[17],广泛种植于亚洲和非洲,喜温暖潮湿的气候、漫射光和弱酸性土壤[18]。干旱、低温、长期辐射、病虫害等非生物胁迫和生物胁迫都会对茶树生长产生不利影响[19-20],其中干旱胁迫通过影响茶树生长的土壤和水分条件,严重降低茶叶的产量和品质[21]。因而研究茶树的抗旱分子机制,挖掘重要的抗性基因,进而通过分子育种技术获得抗性优良品种尤为重要。本研究基于茶树基因组数据库,鉴定获得9个茶树基因家族成员,分析CsSRO家族成员间的系统发育关系、外显子-内含子结构分布及保守基序的组成,并进一步分析9个基因在茶树组织、干旱胁迫和外源ABA处理下的表达模式。笔者旨在通过对茶树基因家族系统的分析,为进一步研究基因介导生理发育过程和应激反应的分子机制奠定基础,为今后茶树遗传改良提供有益的依据。
1 材料与方法
1.1 材料处理
试验在茶学福建省高校重点实验室进行,试验材料选取生长良好、长势一致的两年生盆栽铁观音茶树。将在(24±2)℃的环境温度下生长的植株转移至溶液(10% PEG-6000)中进行干旱处理。将新鲜制备的100 μmol·L-1ABA溶液分别喷洒在不同处理植株的叶片上进行激素处理。收集两种处理6、12、24、48 h后和对照(0 h未处理)的第二叶。每个处理3次重复,锡箔纸包裹标记,液氮速冻后保存于–80℃冰箱备用。
1.2 茶树SRO基因家族成员的鉴定及序列分析
首先下载茶树基因组数据库中的蛋白序列(http://tpia.teaplant.org)[22],从Pfam数据库(https://pfam.xfam.org)下载SRO蛋白特征结构域PARP(PS51059)、RST(PF12174)和WWE(PS50918)的隐马尔可夫模型,并使用HMMER软件进行鉴定[23]。此外,为了进一步验证所鉴定的茶树基因家族,利用SMARAT(http://smart.embl-heidelberg.de)网站和NCBI的Blast-P比对确保上述候选基因至少含有1个PARP催化中心和C-末端的RST结构域。利用ExPASy(http://www.expasy. org)网站分析茶树SRO蛋白的理化性质,在线工具WOLF PSORT(https://wolfpsort.hgc.jp)进行亚细胞定位预测。
1.3 茶树SRO蛋白保守结构域及保守基序分析
利用EBL-EBI(http://www.ebi.ac.uk/interpro/ search/sequence-search)分析氨基酸序列的蛋白质保守区,并用IBS 1.0软件绘制蛋白保守结构域图,使用MEME工具(http://meme- suite.org/tools/meme)对茶树SRO蛋白的保守基序进行分析[24],设置基序数量参数为5个,其余为默认。
1.4 茶树SRO基因结构及系统进化树分析
从茶树基因组数据库中下载茶树外显子和内含子分布数据的存储文件,利用GSDS2.0(http://gsds.cbi.pku.edu.cn)绘制基因结构分布图[25]。从TAIR下载了拟南芥RCD1和SRO1—5的蛋白序列(https://www.Arabidopsis. org),与茶树SRO家族成员一起用ClustalW默认设置对氨基酸序列进行多重序列比对, 并用MEGA 7.0软件进行系统进化树构建,校验参数Boostrap重复为1 000次,其他参数均为默认值[26]。
1.5 茶树SRO基因启动子顺式元件分析
从茶树基因组中提取基因转录起始位置上游2 000 bp的序列进行查找启动子顺式作用元件,在PlantCARE数据库(http://bioinformatics.psb.ugent.be/webtools/plantcare/html)进行预测[27]。
1.6 茶树SRO基因组织特异性及胁迫表达分析
在NCBI SRA(Sequence Read Archive)数据库上下载茶树不同组织的转录组数据,包括顶芽、嫩叶、成熟叶、老叶、花、茎、根(NCBI登录号:SRP056466)[22]。使用TopHat2软件将转录组所有可用读数映射到茶树基因组[28],然后通过HTseq软件计算基因FPKM值表达水平[29],最后对各转录组基因的FPKM值进行归一化处理,使用HemI 1.0软件制作热图。
1.7 RNA提取和实时荧光定量PCR
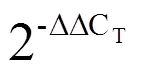
2 结果与分析
2.1 茶树SRO基因家族的鉴定与序列分析
经过分析和验证,最终获得9个家族成员。通过与拟南芥家族成员的同源性进行命名,分别为—和—。基因编码序列长度为489~2 622 bp,编码氨基酸数目为162~873。通过ExPASy软件包对蛋白序列进行分析发现,茶树SRO蛋白家族分子量在17.54~97.78 kDa,等电点为6.07~9.07,CsRCD2属于疏水性蛋白,其他属于亲水性蛋白,CsSROs全部为碱性蛋白。亚细胞预测定位大部分CsSROs(6个)定位于细胞核,CsRCD2定位于细胞质,CsSRO1和CsSRO 5定位于叶绿体(表2)。

表2 茶树CsSRO基因家族的序列特征
2.2 CsSRO家族蛋白结构域组成及保守基序分析
利用EBL-EBI在线分析,发现9个茶树SRO蛋白成员皆含有PARP催化中心和C-末端的RST结构域,而CsRCD4不仅包含PARP和RST结构域,还含有N端的WWE结构域。由于蛋白质的氨基酸序列长度不同,其保守结构域的位置也不同(图1)。CsRCD1、CsRCD3、CsSRO2和CsSRO5的PARP和RST结构域分别位于第220~497个氨基酸和447~586个氨基酸,CsRCD2、CsSRO3和CsSRO4的PARP和RST结构域分别位于第1~302个氨基酸和243~364个氨基酸,CsSRO1的PARP和RST结构域分别位于第2~122个氨基酸和116~162个氨基酸,CsRCD4的WWE、PARP和RST结构域分别位于第78~153个氨基酸、438~658个氨基酸和707~771个氨基酸。
在MEME工具中进行预测发现了茶树SRO家族的5个保守基序(图2)。其中,基序2和基序5高度保守,存在于每个CsSRO的氨基酸序列中,提示这两个基序在SROs基因家族中具有重要作用,通常位于CsSRO多肽的中心和末端附近,具有典型的PARP和RST结构域。基序4只在CsRCD1—4中发现,CsSRO1—5缺少基序4。CsSRO1、CsSRO3还分别缺少基序1和3。结合CsSROs的结构域位置分析可以看出,基序1、基序3和基序4位于PARP保守结构域中。
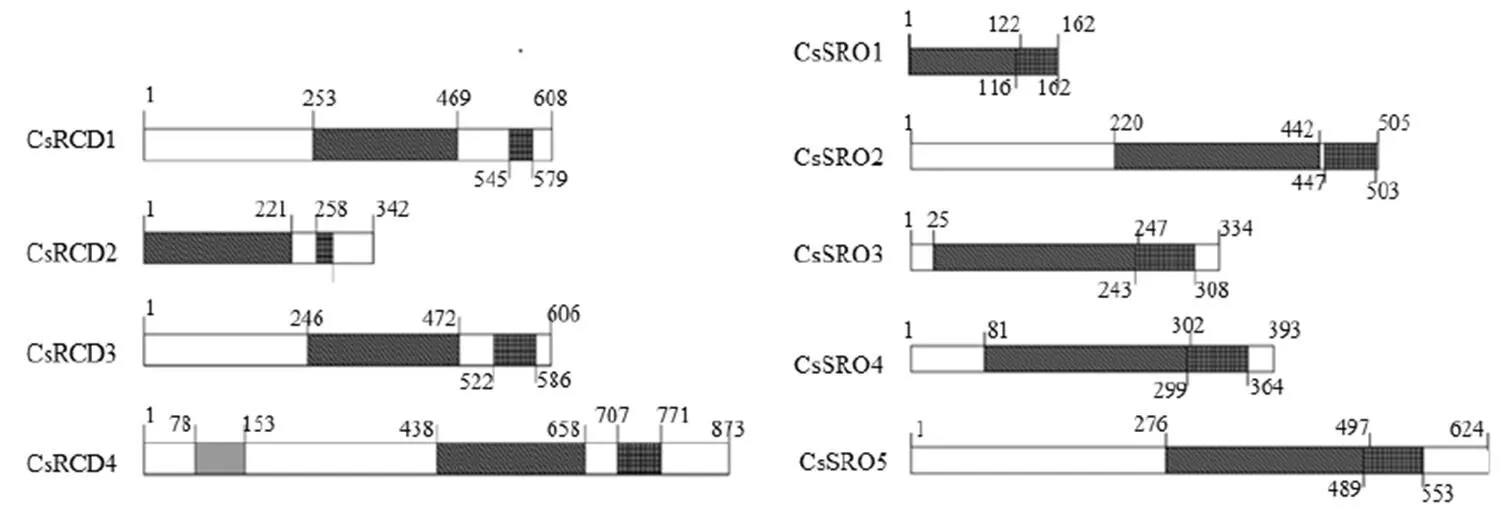
注:灰色区域、黑色斜纹区域和黑色网格区域分别表示CsSROs 蛋白的WWE、PARP 和RST 结构域

图2 茶树SRO 家族保守基序和基序logo 分析
2.3 CsSRO家族进化树及基因结构分析
将茶树与拟南芥SRO家族蛋白序列使用ClustalW进行多重序列比对,然后进行系统进化树构建(图3)。结果显示,茶树SRO可以分为3组,CsRCD1—4与AtRCD1、AtSRO1聚为第Ι组,CsSRO1、CsSRO2聚为第Ⅱ组,CsSRO3—5与AtSRO2—5聚为第Ⅲ组,其中第Ⅰ组和第Ⅲ组同时包含了茶树和拟南芥蛋白。第Ⅰ组中,CsRCD1、CsRCD2和CsRCD3同源性较高,第Ⅲ组中,CsSRO4和CsSRO5同源性较高,相似性均在85%以上。CsRCD1和CsRCD2、CsSRO1和CsSRO2的相似性均在99%以上,可能为旁系同源基因。
利用基因家族成员外显子-内含子结构绘制图谱(图4)。家族基因中基因组序列最长的是,长度超过39 kbp;9个基因的外显子数范围在4到9之间。大多数基因(、3、、和)有5个外显子,、分别有4个和6个,而和有9个外显子。由此发现,相同组的基因外显子数目比较接近。
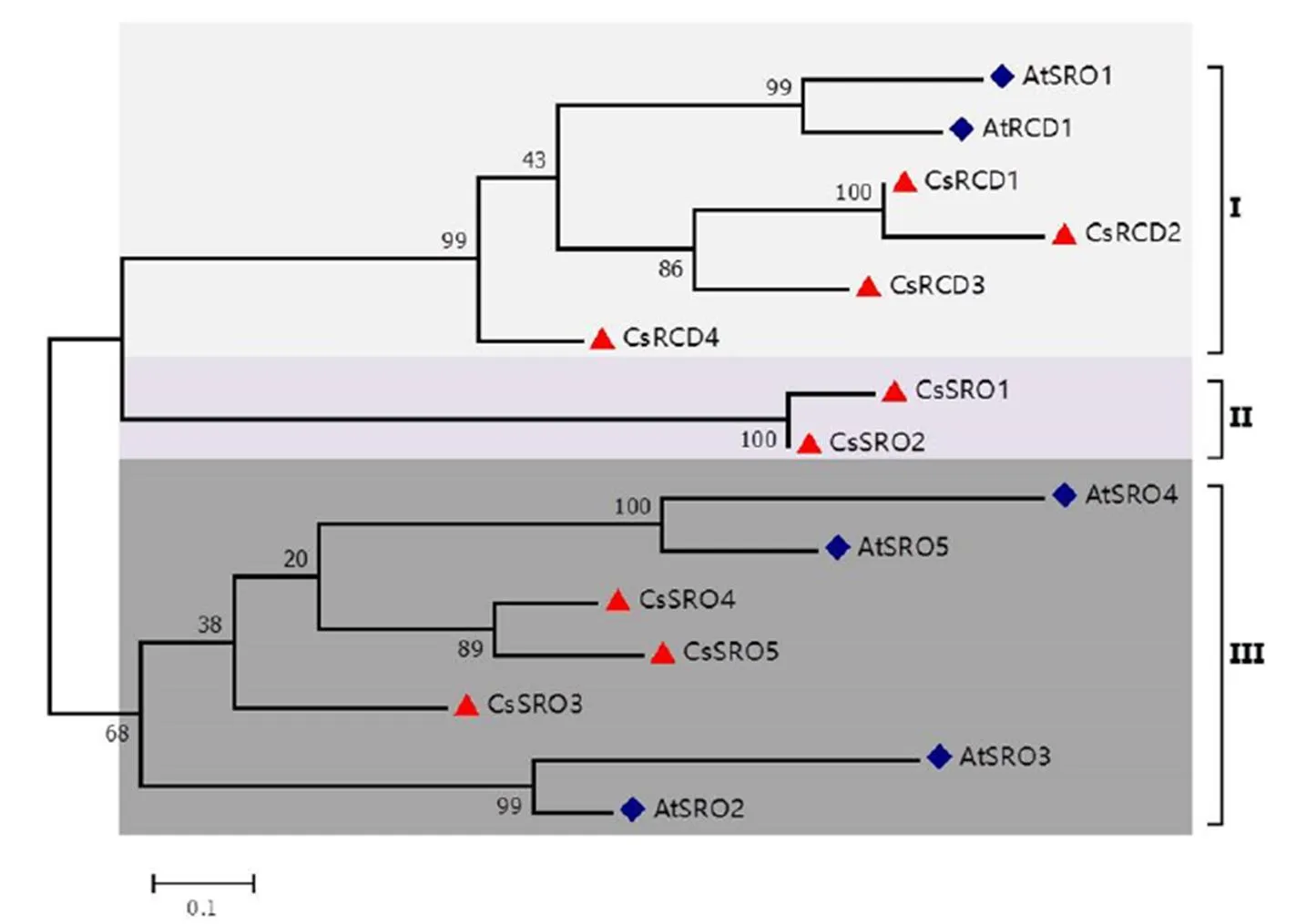
图3 茶树、拟南芥SRO蛋白的系统进化树

图4 茶树SRO基因结构分析
2.4 CsSRO基因启动子顺式元件分析
为了研究基因对各种信号因子的应答作用,用其转录起始位置上游2 000 bp的序列查找各类顺式作用元件,发现有许多光信号元件(MRE、box-4、TCT-motif等22种)、生长发育相关的元件(MBST、RY-element、CAT-box等8种),还有很多激素和胁迫响应元件。重点对基因启动子中与激素和胁迫响应相关的顺式作用元件进行分析并做图(图5),由图中可知,基因启动子含有12种响应激素和胁迫的顺式调控元件,主要包括脱落酸(ABRE,12个)、赤霉素(P-box,2个;GARE-motif,3个;TATC-box,2个)、茉莉酸甲酯(CGTCA-motif,7个;TGACG-motif,7个)、水杨酸(TCA-element,3个)、生长素(TGA-element,3个)等激素响应元件,还有厌氧诱导(ARE,24个)、防御和应激响应元件(TC-rich,3个)、低温响应元件(LTR,3个)、参与干旱诱导的MYB结合位点(MBS,3个)等胁迫响应顺式作用元件。这暗示了茶树SRO家族在茶树生长发育及多种激素和胁迫中的潜在作用。

图5 CsSRO基因启动子区顺式元件分析
2.5 基于转录组数据的CsSRO基因在茶树不同组织中的表达模式
为了挖掘家族在茶树生长发育过程的潜在功能,从网上下载茶树8个组织(顶芽、嫩叶、成熟叶、老叶、根、茎、花、果)的转录组,分析9个基因在茶树8个组织中的表达模式。在组织中不表达,其他成员在各个组织均有表达,因此只对另外8个基因重点分析(图6)。和在各个组织中的表达水平较低,而、在各个组织中均高表达。和在6个组织中较高表达,分别在花和根中的表达量最高。成熟叶和根中的表达量最高。总的来说,大多数基因在根和成熟叶中较高表达(FPKM>14),推测可能在根和叶中发挥重要生物学功能。
2.6 CsSRO基因在干旱胁迫和外源ABA处理下的表达模式
通过荧光定量技术对基因在干旱胁迫和外源ABA处理0、6、12、24、48 h后的表达水平进行分析,0 h的处理为对照(图7)。干旱处理下,9个茶树基因均被诱导表达,但表达水平在处理的不同时间点有一定差异。在4个时间点均被下调表达,24 h时表达量最低。的表达量在12 h时达到峰值,24 h时下降到控制水平,48 h时表达量又上升,表达量又下降。、、、、在4个时间点均以不同程度被上调表达,峰值表达量分别是0 h的4.9、3.4、2.7、398.4、27.9和5.6倍。ABA处理下,大多数都存在明显的响应,变化趋势不尽相同。的表达量在4个时间点的变化不显著。、、、在4个时间点均被抑制表达。在不同时间点均被上调表达,的相对表达量在6 h时上升随后逐渐下降到控制水平以下。和最高可上调表达3倍和1.8倍。
3 讨论
SRO蛋白家族在陆地植物中高度保守[6],其生物学功能及分子调控机制受到越来越多的关注,然而,的功能在茶树中还是未知的。本研究从茶树基因组中鉴定出的9个茶树基因均具有PARP和RST等特征保守结构域,其中CsRCD4具有WWE结构域,分别起重要功能作用[14]。通过与拟南芥SRO家族进行系统发育分析发现,家族基因中可能含有旁系同源基因,说明在长期进化过程中发生了基因重复事件[31]。在拟南芥中,根据SRO蛋白家族是否具有N端WWE结构域将其分为两类,CsRCD1—4与具有N端WWE结构域的AtRCD1、AtSRO1聚为第Ι组,CsRCD4和AtRCD1、AtSRO1一样同时具有PARP、RST和WWE域,CsRCD1—3缺失WWE域,但CsRCD1—3与AtRCD1、AtSRO1系统进化树亲缘关系更近,CsRCD1、CsRCD2和CsRCD3之间同源性也更高,这表明PARP和RST域在茶树中比WWE域保守。CsSROs基因亚组间基序分布有一定差异,表明不同组的CsSRO基因在功能上有所差异[32],例如,基序4仅在Ι组中发现,Ⅱ和Ⅲ组都缺少基序4,这表明Ι组成员较于其他类群具有特殊的功能,CsSRO1和CsSRO3还分别缺少基序1和3,这可能是在组织中不表达、强烈响应干旱胁迫的原因之一。对外显子和内含子的研究有助于了解基因的结构和功能上的差异[33],相同组的基因外显子数目很接近,例如Ι组中的CsRCD1、CsRCD2和CsRCD3,Ⅱ组中的CsSRO1和CsSRO2,Ⅲ组中的CsSRO3和CsSRO4,因此,大多数基因呈现保守的基因结构,支持密切的进化关系。
基因在植物不同组织中的表达水平与其生长发育调控密切相关。有研究表明大部分苹果基因在根、茎和花中的表达量较高[15],玉米家族基因在根系的表达量显著高于其他组织[5]。与之相似,和分别在花和根中的表达水平显著高于其他组织,暗示可能在茶树花器官发育发挥重要作用,在根系发育中发挥作用。在8个茶树组织中均高表达,提示其具有重要作用,可能参与茶树发育过程中多种生理生化的转录调控过程[31]。有趣的是,和均在根和成熟叶中的表达量较高。根部是植物吸收水分和矿物质的重要部位,叶片蒸腾作用造成水分损失[34]。推测可能通过影响茶树根系吸水能力和叶片蒸腾作用,增强茶树耐干旱能力。
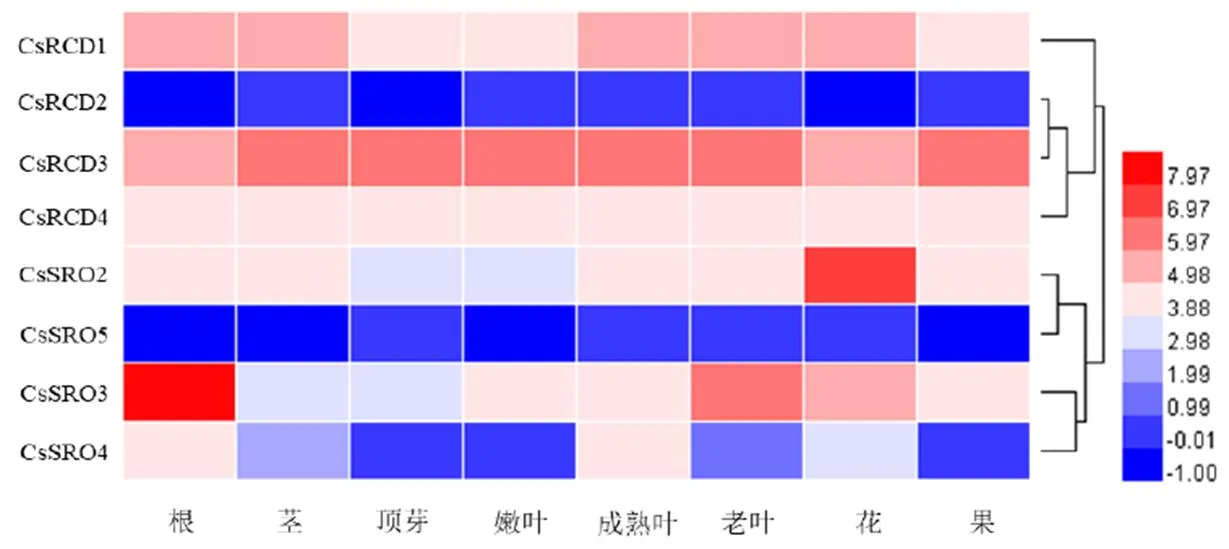
注:颜色刻度表示log2转换后的值,红色代表高表达,蓝色代表低表达
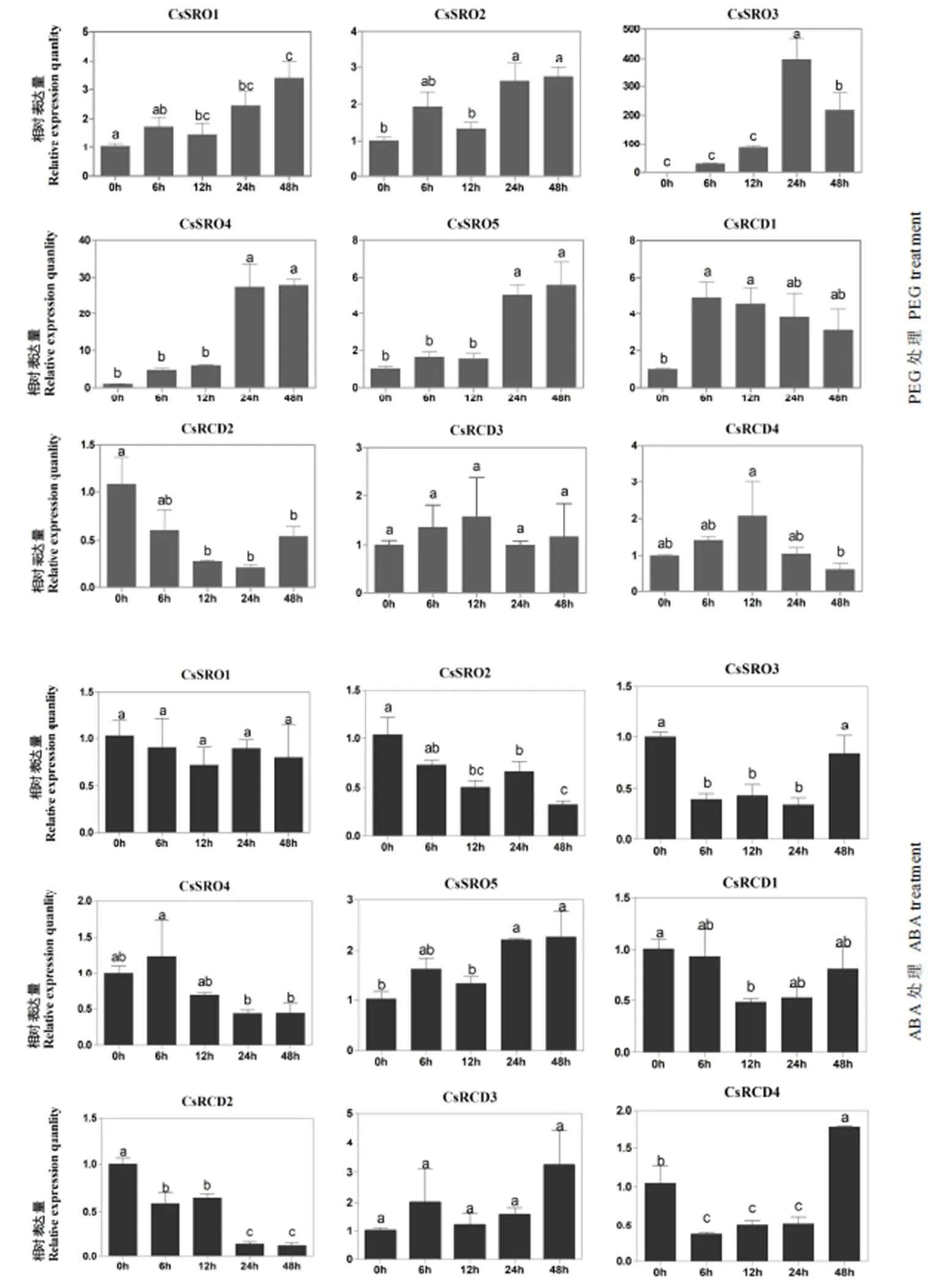
图7 CsSROs基因在干旱胁迫和脱落酸处理下的表达分析
为了研究茶树基因在干旱胁迫下的响应情况,本研究分别采用PEG和外源ABA处理茶树叶片,荧光定量分析结果证实在干旱和ABA处理下均有表达且表达受到不同程度的诱导。拟南芥中,定位于细胞核的与转录因子DREB2A相互作用,参与植物ABA依赖的干旱反应[14]。亚细胞预测定位于细胞核的与具有较高的同源性,在干旱胁迫中上调表达,其中在ABA处理下显著上调表达,可能具有类似的生物学功能。玉米中,和在干旱条件下显著上调表达[5]。、在干旱处理不同时间点下最高可以分别上调4.9、3.4、2.7、398.4、27.9、5.6倍,表明这6个成员尤其是对干旱胁迫具有较高的敏感性。其中,在ABA处理不同时间点均被诱导下调表达。与茶树相似,异位表达的苹果降低转基因幼苗对外源ABA的敏感,并且耐受干旱胁迫[15],推测可能参与ABA信号途径负调控干旱反应。在基因启动子区域发现很多与脱落酸及干旱胁迫响应调控相关的顺式元件,也预示着基因家族有可能通过激素信号转导途径响应干旱胁迫。
本研究在茶树全基因组范围内鉴定获得基因家族,并分析其结构和功能,推测基因与茶树的生长发育和抗旱响应密切相关,为深入了解茶树中该基因家族成员的功能提供了参考和一定的研究基础。
[1] 岳川, 曹红利, 郝心愿, 等. 茶树基因的克隆及其表达分析[J]. 茶叶科学, 2017, 37(4): 399-410.
[2] Liu S, Liu S, Wang M, et al. A wheatgene enhances seedling growth and abiotic stress resistance by modulating redox homeostasis and maintaining genomic integrity [J]. The Plant Cell, 2014, 26(1): 164-180.
[3] You J, Zong W, Du H, et al. A special member of the rice SRO family,, mediates responses to multiple abiotic stresses through interaction with various transcription factors [J]. Plant Molecular Biology, 2014, 84(6): 693-705.
[4] 吕有军, 杨卫军, 赵兰杰, 等. 陆地棉基因家族的鉴定及表达分析[J]. 作物学报, 2017, 43(10): 1468-1479.
[5] 赵秋芳, 马海洋, 贾利强, 等. 玉米基因家族的鉴定及表达分析[J]. 中国农业科学, 2018, 51(15): 196-206.
[6] Jaspers P, Overmyer K, Wrzaczek M, et al. The RST and PARP-like domain containing SRO protein family: analysis of protein structure, function and conservation in land plants [J]. BMC Genomics, 2010, 11: 170. DOI: 10.1186/1471-2164-11-170.
[7] Katiyar-Agarwal S, Zhu J, Kim K, et al. The plasma membrane Na+/H+antiporter SOS1 interacts with RCD1 and functions in oxidative stress tolerance in[J]. Proceedings of the National Academy of Sciences, 2007, 103(49): 18816-18821.
[8] Ahlfors R, Overmyer K, Jaspers P, et al. Arabidopsis radical-induced cell death 1 belongs to the WWE protein-protein interaction domain protein family and modulates abscisic acid, ethylene and methyl jasmonate responses [J]. Plant Cell, 2004, 16(7): 1925-1937.
[9] Vainonen J P, Jaspers P, Wrzaczek M, et al. RCD1-DREB2A interaction in leaf senescence and stress responses in[J]. Biochemical Journal, 2012, 442(3): 573-581.
[10] Teotia S, Lamb RS. The paralogous genesandhave partially redundant functions during Arabidopsis development [J]. Plant Physiology, 2009, 151(1): 180-198.
[11] Jaspers P, Blomster T, Brosche M, et al. Unequally redundant RCD1 and SRO1 mediate stress and developmental responses and interact with transcription factors [J]. The Plant Journal, 2009, 60(2): 268-279.
[12] Zhao X, Gao L, Jin P, et al. The similar to RCD-one 1 protein SRO1 interacts with GPX3 and functions in plant tolerance of mercury stress [J]. Bioscience Biotechnology and Biochemistry, 2017, 82(1): 1-7.
[13] Babajani G, Effendy J, Plant AL.increases salt tolerance and is a member of the—gene family of tomato [J]. Plant Science, 2009, 176(2): 214-222.
[14] 李保珠, 赵翔, 赵孝亮, 等. 拟南芥SRO蛋白家族的结构及功能分析[J]. 遗传, 2013, 35(10): 1189-1197.
[15] Li H, Li R, Qu F, et al. Identification of thegene family in apples () with a functional characterization of[J]. Tree Genetics & Genomes, 2017, 13(5): 94. DOI: 10.1007/s11295-018-1242-4.
[16] You J, Zong W, Li X, et al. The SNAC1-targeted genemodulates stomatal closure and oxidative stress tolerance by regulating hydrogen peroxide in rice [J]. Journal of Experimental Botany, 2013, 64(2): 569-583.
[17] Wang W, Xin H, Wang M, et al. Transcriptomic analysis reveals the molecular mechanisms of drought-stress-induced decreases inleaf quality [J]. Frontiers in Plant Science, 2016, 7: 385. DOI: 10.3389/fpls.2016.00385.
[18] Zhou Y, Liu Y, Wang S, et al. Molecular cloning and characterization of galactinol synthases inwith different responses to biotic and abiotic stressors [J]. Journal of Agricultural and Food Chemistry, 2017, 65(13): 2751-2759.
[19] Hou Y, Wu A, He Y, et al. Genome-wide characterization of the basic leucine zipper transcription factors in[J]. Tree Genetics & Genomes, 2018, 14(2): 27. DOI: 10.1007/s11295-018-1242-4.
[20] Liu L, Li Y, She G, et al. Metabolite profiling and transcriptomic analyses reveal an essential role of UVR8-mediated signal transduction pathway in regulating flavonoid biosynthesis in tea plants () in response to shading [J]. BMC Plant Biology, 2018, 18(1): 233. DOI: 10.1186/s12870-018-1440-0.
[21] Zhang Q, Cai M, Yu X, et al. Transcriptome dynamics ofin response to continuous salinity and drought stress [J]. Tree Genetics & Genomes, 2017, 13(4): 1-17.
[22] Wei C, Yang H, Wang S, et al. Draft genome sequence ofvar. sinensis provides insights into the evolution of the tea genome and tea quality [J]. Proceedings of the National Academy of Sciences, 2018, 115(18): 4151-4158.
[23] Xia EH, Zhang HB, Sheng J, et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis [J]. Mol Plant, 2017, 10(6): 866-877.
[24] Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching [J]. Nucleic Acids Research, 2009, 37: 202-208.
[25] Hu B, Jin J, Guo A, et al. GSDS 2.0: an upgraded gene feature visualization server [J]. Bioinformatics, 2015, 31(8): 1296-1297.
[26] Hall B G. Building phylogenetic trees from molecular data with MEGA [J]. Molecular Biology and Evolution, 2013, 30(5): 1229-1235.
[27] Lescot M. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences [J]. Nucleic Acids Research, 2002, 30(1): 325-327.
[28] Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks [J]. Nature Protocols, 2012, 7(3): 562-578.
[29] Anders S, Pyl P T, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data [J]. Bioinformatics, 2015, 31(2): 166-169.
[30] Livak K J, Schmittgen T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method [J]. Methods, 2001, 25(4): 402-408.
[31] 魏瑞敏, 郑井元, 刘峰, 等. 辣椒家族基因的鉴定与表达分析[J]. 园艺学报, 2018, 45(8): 1535-1550.
[32] Wang YX , Liu ZW , Wu ZJ, et al. Genome-wide identification and expression analysis of GRAS family transcription factors in tea plant () [J]. Scientific Reports, 2018, 8(1): 3949. DOI: 10.1038/s41598-018-22275-z.
[33] Xu G, Guo C, Shan H, et al. Divergence of duplicate genes in exon-intron structure [J]. Proceedings of the National Academy of Sciences, 2012, 109(4): 1187-1192.
[34] 岳川, 曹红利, 王赞, 等. 茶树水通道蛋白基因的克隆与表达分析[J]. 西北植物学报, 2018, 38(8): 1419-1427.
Genome-wide Identification and Expression Analysis ofGene Family in
GUO Yongchun, WANG Pengjie, CHEN Di, ZHENG Yucheng, CHEN Xuejin,YE Naixing*
College of Horticulture, Fujian Agriculture and Forestry University/Key Laboratory of Tea Science at Universities in Fujian, Fuzhou 350002, China
(Similar to rcd one) are plant-specific gene families. In this study, 9gene family members were identified from tea tree genome by bioinformatics method and named asandrespectively. All coding proteins of the 9genes have characteristic structural domains PARP and RST, and have similar conserved motifs. The CsSRO genes were divided into 3 groups based on phylogenetic tree analysis, with the groupΙcontaining CsRCD1—4, the groupⅡcontaining CsSRO1, CsSRO2 and the group Ⅲcontaining CsSRO3—5. Gene structure analysis shows that this gene family contained 4 to 9 exons. Analysis of transcriptome data from 8 tea tree tissues shows thatmight play an important role in different developmental stages of tea plants. Mostgenes were highly expressed in roots and mature leaves. Upstream promoter region analysis found a large number of-acting elements closely related to plant development, hormones and stress response. Further expression analysis shows that 9genes were induced by drought and abscission acid treatments, suggestinggenes may be closely related to drought resistance.
,, phylogeny analysis, stress
S571.1;S154.1
A
1000-369X(2019)04-392-11
2019-02-11
2019-03-26
福建省“2011协同创新中心”中国乌龙茶产业协同创新中心专项(闽教科〔2015〕75号)、国家现代农业(茶叶)产业技术体系建设专项资金项目(CARS-19)、福建农林大学科技创新专项基金项目(CXZX2016117、CXZX2017181)
郭永春,女,硕士研究生,主要从事茶树栽培育种与生物技术研究方面的研究。*通信作者:ynxtea@126.com
