成人型神经元核内包涵体病2例临床及病理研究
2019-08-16董明睿洪道俊邵自强彭丹涛
王 璐 ,董明睿 ,洪道俊 ,袁 云 ,汪 伟 ,邵自强 ,彭丹涛
(1.中日友好医院 神经科,北京 100029;2.北京大学人民医院 神经内科,北京 100044;3.北京大学第一医院 神经内科,北京 100034)
神经元核内包涵体病(neuronal intranuclear inclusion disease,NIID)是一种慢性进展的神经系统退行性疾病,上世纪60~80年代Lindenberg、Sung等学者先后提出了NIID的诊断[1,2]。根据发病年龄该病分为婴儿型,青少年型和成人型,成年型则分为家族型和散发型[3]。早期欧美国家的研究中,通过尸检,直肠、神经活检诊断的患者多数为婴儿期或青少年期起病,以大脑皮层功能障碍和锥体外系症状为主要表现[4]。近10年来随着皮肤活体组织检查在NIID诊断中的价值被证实,日本学者报道了60余例NIID患者,多数为成年起病,临床表现复杂多变,可出现痴呆、自主神经功能障碍、锥体外系病变、周围神经病、小脑受累、癫痫发作、精神行为异常等症状[5]。国内对该病的研究起步较晚,经病理证实的成人型NIID国内仅有1例报道[6],我们报道2例确诊患者,并就其临床和病理特点进行初步探讨。
1 临床资料
例1女,61岁。间断意识障碍,伴发热、呕吐半年余;再发半个月。半年前患者高热后呼之不应,抗感染治疗后体温正常、意识转清(类似症状半个月内发生2次),病程中反复恶心、呕吐,醒后自发言语少,伴左侧口角流涎,左上肢活动不利,经过家中康复训练后,可扶物行走。
半个月前患者恶心、呕吐2d,呼之不应,体温38℃,血压99/69mmHg,昏迷,双眼向右侧凝视,双侧瞳孔等大等圆,对光反射存在;右侧肢体疼痛刺激可躲避;双侧巴氏征阳性。再次抗感染治疗后,患者体温正常,查体神清,言语少,定向力差;眼位居中;左侧肢体肌力3级以上,右侧肢体肌力4级。既往高血压病史,血压波动明显;时有恶心、呕吐,行胃镜检查无异常;尿潴留,泌尿系感染,留置尿管;抑郁症,常有畏惧、幻视症状。可疑阳性家族史(图1)。血常规白细胞总数正常,中性粒细胞百分比76.9%略高。腰穿脑脊液常规生化、病原学及自身免疫抗体谱、副肿瘤抗体谱未见异常、脑脊液14-3-3蛋白阴性。2017年头颅MRI(图2A-D,封二):FLAIR序列双侧额顶颞枕叶及脑岛皮层下白质异常信号影,右侧颞叶斑片状高信号;弥散加权成像(diffusion weighted imaging,DWI)可见双侧额、顶、颞叶沿皮髓质交界处高信号。半年余后头颅MRI(图2E-H,封二):白质病变较前略加重,右侧颞叶异常信号影变小;DWI可见额、顶及颞叶皮髓交界高信号。头颈CTA示头颈动脉硬化。肌电图提示多发周围神经病变。脑电图中度异常,可见各导联弥漫持续中波幅1~1.5次/s的复形慢波。患者胃管、尿管置管状态,多次住院予抗感染(细菌、病毒),补液、补钾、护胃、控制血压(血压波动)及营养支持治疗。
例2女,60岁。头晕伴恶心、呕吐2周。2周前患者空腹时无明显诱因出现头晕,头部昏沉,伴恶心、呕吐,呕吐物为胃内容物,呕吐共6~7次,与体位及活动无关,加重及缓解方式不明,无自身旋转感,无肢体无力及抽搐,无面部及肢体麻木,无声音嘶哑,无饮水呛咳及吞咽困难,无意识障碍,无二便障碍。于我院急查头颅CT示:“双侧基底节区、放射冠区、半卵圆中心多发脑梗塞;脑白质变性”;予“抗血小板、调脂、改善循环、止晕”等治疗,后患者症状较前好转。既往史:高血压病史;视网膜色素变性,近1年视力下降较明显,目前仅有光感。否认家族史。查体:神清语利,高级皮层功能正常;颅神经检查未见明显异常;四肢肌力5级,双侧肌张力大致正常。双侧深、浅感觉对称存在。双侧腱反射减低,双侧病理征阴性。双侧共济稳准,Romberg征阴性。腰穿脑脊液常规生化、病原学及自身免疫抗体谱、副肿瘤抗体谱未见异常。头颅MRI(图2I-L,封二):FLAIR序列双侧额顶颞及胼胝体压部白质异常信号影;DWI可见双侧额、顶、颞叶沿皮髓质交界处高信号。头颈CTA示双侧颈内动脉虹吸段钙化斑块,右大脑中动脉M1段轻度狭窄。肌电图示周围神经病变。入院期间予改善循环、补液、止晕及营养神经治疗。

图1 例1家系图

2 病理检查方法
2例患者的皮肤活检标本均经开放的活检手术方法获得,活检部位为左小腿(距外踝约10cm处)。新鲜的皮肤活检标本分为2部分,一部分经液氮冷却的异戊烷速冻,制成8μm冰冻切片,行苏木精-伊红(HE)、刚果红染色,及免疫组织化学染色检测p62蛋白表达情况;另一部分标本经3%戊二醛固定,Epon812包埋,制成1μm半薄切片,甲苯胺蓝染色定位,制成超薄切片,铅-铀双染色,日立H-800透射电镜下观察。
3 病理诊断结果
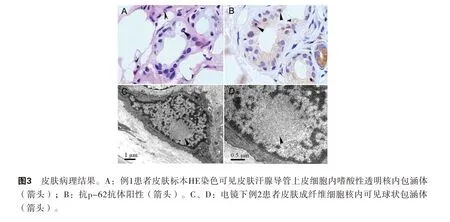
例1患者皮肤病理改变:HE染色可见部分汗腺导管上皮细胞核内存在嗜酸性核内包涵体,免疫组织化学染色可见包涵体p62阳性(图3A、B,封二)。刚果红未发现阳性物质沉积。
例2患者HE染色未见汗腺腺体、脂肪细胞细胞核内类圆形包涵体;刚果红未发现阳性物质沉积;免疫组织化学染色未见p62阳性物质。电镜可见皮肤成纤维细胞核内可见球状、浅染的短丝状包涵体结构,大小约 1.5μm×2μm(图3C、D,封二);汗腺导管部和分泌部上皮细胞,血管内皮细胞内均未见异常沉积物。
4 讨论
4.1 诊断依据
1968年Lindenberg首次提出NIID的诊断[1],之后数十年中研究者们通过尸检、直肠及神经活检发现嗜酸性透明包涵体,报道了40余例NIID患者[3,4,7]。2011 年 Sone 等报道采用皮肤活检标本可诊断NIID[8],2014年再次提出头颅DWI显示皮髓质交界处特征性的高信号改变为诊断NIID的另一重要线索[9]。目前认为皮肤活检结合头颅DWI特征改变是该病的主要诊断依据。2018年关于肾脏活检早于临床症状和头颅DWI特征改变12年即可发现嗜酸性包涵体的报道[10],进一步提高了病理诊断的价值。本研究中2例患者皮肤活组织检查分别在光镜和电镜下发现核内包涵体,结合头颅MRI特征性的弥散像皮髓质交界高信号,可确诊为神经元核内包涵体病。
4.2 临床表现
本研究中2例患者均为60岁起病,符合成人型神经元核内包涵体病的起病年龄;例1患者存在可疑阳性家族史,考虑为家族型NIID,例2患者为散发型NIID。2例患者均有发作性消化道症状(反复恶心、呕吐),其中较重的1例出现间断意识障碍、认知障碍、肢体无力、膀胱功能异常;较轻的患者表现为反复头晕发作,进一步验证了NIID的临床异质性,临床工作中需要提高警惕性,减少漏诊[5]。国内陈为安教授等报道了1例61岁女性患者[6],主要表现为发作性脑病、发作性消化道症状、缓慢进展的智能减退、自主神经损害;其发病年龄和临床表现与例1患者类似,尽管缺乏国内的大宗报道,但我们推测发作性消化道症状可能是我国患者的主要临床表现之一。家族型的报道中以亚急性脑炎为主要表现的病例少见,痴呆患者常伴轻微的自主神经功能紊乱;例1反复出现意识障碍伴发热、呕吐等亚急性脑炎症状,且自主神经功能障碍早于肢体无力数年出现,与既往报道不一致[5]。例2较以往散发型的报道,临床症状较轻,仅出现反复头晕伴恶心、呕吐,并未出现痴呆的表现,推测可能与病程较短有关。此外,本研究中2例患者的肌电图结果均提示了周围神经损害,高级皮层功能、周围神经系统及自主神经系统同时受累,有助于在临床工作中鉴别该病与其他神经系统变性病。
4.3 头颅DWI特征表现
头颅DWI显示皮髓质交界处特征性的高信号是该病特异性的影像学表现,也称作皮质下绸带征(subcortical lace sign)。DWI高信号局限于皮髓交界区,随着疾病进展逐步在额、顶、颞、枕叶皮层扩展,但即使在后期也不会深入到脑白质,可以作为诊断标记物,尚未见于其他疾病的报道[9,11]。有报道中T2及FLAIR序列出现白质高信号,类似白质脑病的表现,弥漫且对称,额叶为著;部分痴呆组中可出现局灶性水肿及强化灶;而侧脑室扩张、小脑萎缩等表现也可见于散发型和家族型患者[12]。本研究中患者头颅MRI表现与病程、临床症状的严重程度相符。例1患者半年后的皮质下绸带征较前无显著变化,额叶的白质病变更为显著;而在发病初期的右颞叶局灶性水肿较半年后明显。病程较短的例2患者的头颅DWI同样出现了皮质下绸带征,前头部的高信号更明显;同时伴胼胝体压部的受累。
4.4 机制探讨
神经元核内包涵体病的病理生理机制尚不明确。神经元核内包涵体(neuronal intranuclear inclusions,NIIs)见于多种神经系统变性病,其形成与泛素-蛋白酶体系统的功能障碍有关[7]。目前无明显证据表明NIIs的形成与神经细胞坏死和神经缺失有关[13],即使在多聚谷氨酰胺疾病的研究中也未发现NIIs致病的直接证据[7]。因此该病尚缺乏特效的病因治疗方法,临床多以对症为主,仅有少数激素冲击治疗的病例报道。
总之,对比国外报道,本研究初步总结了国内NIID患者的临床及皮肤病理特征,进一步证实了皮肤活检的诊断价值。
