黄芩素的结构修饰以及抗肿瘤活性研究
2019-08-02陈党辉邓雁如牟佳佳
陈党辉,邓雁如,牟佳佳,贾 妍
天津中医药大学中药学院,天津 301617
黄芩素是传统中药黄芩中的有效成分,具有神经元保护、抗氧化、抗菌、抗病毒、抗过敏、抗炎、抗肿瘤等多种生理活性[1]。近年来,对黄芩素抗肿瘤作用的研究报道逐渐增多。早在1999年,Tomoo等人[2]在研究黄芩素对鼠肝脏肿瘤细胞的抗增殖作用时,就发现黄芩素显示出了较强的抑制肝肿瘤细胞增殖的活性。黄芩素对很多肿瘤细胞均有较强的抑制作用,且因肿瘤细胞种类不同,其抑制作用强弱不同[3]。细胞周期蛋白依赖性激酶1(CDK1)协调从G2期向M期的转变,并且由于癌细胞通常表现出增强的CDK1活性,因此将其作为肿瘤特异性抗癌靶标[4]。黄芩素是一种天然的CDK1抑制剂[5],2009年,美国纽约大学西奈山医学院David[6]报道了黄芩素诱导人肝癌化HepG2细胞调亡,与选择性抑制CDK1,CDC25B等表达有关[5]。在天然黄酮中黄芩素是最有潜力的选择性的CDK1抑制剂[7],其体外抗细胞增殖活性IC50为25~75 μmol/L[8]。有望开发成新型的 CDK1抑制剂类抗癌药物[5]。由于该化合物在植物中广泛存在,对正常组织低毒甚至无毒,同时黄芩素属多元酚类抗氧剂,具有抑制细胞增殖,能选择性诱导肿瘤细胞调亡和保护正常细胞避免发炎坏死作用[5,9]。其抗肿瘤作用的研究工作仍在不断进行,是今后新药开发研究中一个值得重视的资源,有较好的开发利用前景[10]。
但是黄芩素结构中存在3个相邻酚羟基,易于形成分子内氢键,造成其亲脂性、亲水性均较差,在临床应用上存在着溶解性差、生物利用度低等缺点[1,11]。因此,为改善黄芩素溶解性、提高抗肿瘤活性,改善其在临床上的应用,以其为先导化合物,利用化学反应对其进行结构修饰引起了广泛关注。其中有机胺类化合物是一个研究热点,李康健等人对黄芩素进行了8位氨基mannich反应,结果显示绝大多数所测化合物对CDK1/cyclinB都显示出了较高的抑制活性,其IC50值在1.05~1.28 μmol/L之间,大约为黄芩素的6倍[3]。因此为进一步改变其理化性质、提高生物利用度、改善活性,本文通过mannich反应在黄芩素A环8位引入胺亚甲基侧链,同时通过酯化反应在其6位(7位)羟基上引入不同的疏水性基团,丰富黄芩素衍生物的结构,通过活性评价进而探讨黄酮类抗肿瘤的构效关系,为开发具有知识产权的抗肿瘤药物奠定物质基础。由于黄芩素结构中有三个酚羟基,因此在进行酯化反应的过程中,酯键发生在哪个羟基上是不清楚的,本文一并对此问题进行研究。
1 实验材料
黄芩素,98%,购自南京泽郎生物科技有限公司;硫代吗啉,98%,购自天津市江天化工技术有限公司;甲醇、37%甲醛、三乙胺、二氯甲烷、四氢呋喃,均为分析纯,购自天津市康科德科技有限公司;苯甲酸、对甲氧基苯甲酸、邻溴苯甲酸、甲基丙烯酸、肉桂酸,纯度均大于98%,购自天津市博迪化工有限公司;N-羟基苯并三氮唑(HOBT),98%,购自damas-beta公司;二环己基碳酰二亚胺(DCC),99%,购自solarbio公司;二甲氨基吡啶、碳酸钾、碘化钾,均为分析纯,购自天津市江天化工技术有限公司;GF254薄层板、200目硅胶,购自青岛海洋化工有限公司;600 M AVIII核磁共振谱仪(瑞士,布鲁克);6540 UHD accurate mass Q-TOF MS高分辨质谱仪(美国,安捷伦);MCF-7(人乳腺癌细胞),购自河北蓝梦生物科技有限公司;CCK-8试剂盒,购自日本同仁化学。
2 实验方法
本文以黄芩素为原料,按图1所示的合成路线,首先通过mannich反应,在8位引入胺亚甲基,然后通过酰基化反应在酚羟基上引入不同的疏水性基团。通过波谱手段与化学手段相结合,对合成目标化合物进行结构确证以及利用CCK-8法进行抗肿瘤活性评价。
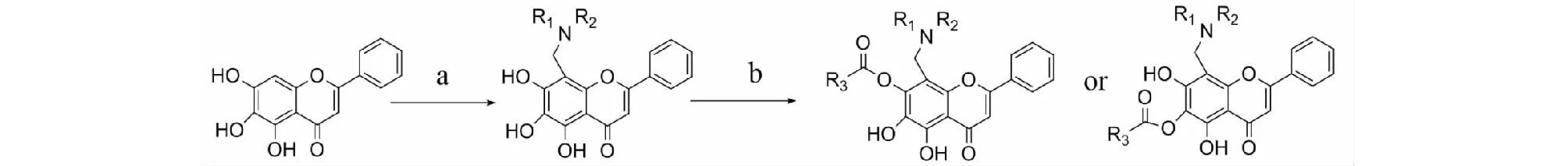
图1 黄芩素衍生物的合成路线Fig.1 Synthesis route of baicalein derivatives注:试剂和条件,(a) HCHO,NHR1R2/MeOH;(b)羧酸,DCC,HOBT/无水THF。Note:Reagents and conditions,(a) HCHO,NHR1R2/MeOH;(b) carboxylic acid,DCC,HOBT/anhydrous THF.
2.1 黄芩素的Mannich碱衍生物的制备
2.1.1 5,6,7-三羟基-2-苯基-8-(硫代吗啉亚甲基)-4H-苯并吡喃-4-酮(化合物1)的制备
参考文献[12],称取黄芩素1.08 g(4 mmol)于150 mL反应瓶中,加入甲醇30 mL作溶剂,在搅拌条件下,依次加入37%的甲醛溶液0.486 mL(6 mmol),硫代吗啉0.483 mL(4.8 mmol),于50 ℃条件下反应,薄层色谱法监测反应进程,直至反应完全,有大量黄色固体析出;将黄色固体抽滤,用少量甲醇洗涤滤饼,将滤饼于真空干燥箱中干燥,称重:1.36 g,产率88.7%;1H NMR (DMSO-d6,600 MHz)δ:12.78 (s,1H ,5-OH),8.09 (dd,J=7.7 Hz,1.4 Hz,2H,H-2′,6′),7.59~7.63 (m,3H,H-3′,4′,5′),6.95 (s,1H,H-3),3.97 (s,2H,H-9),2.87 (s,4H,H-10,13),2.67 (s,4H,H-11,12);13C NMR (DMSO-d6,150 MHz) δ:182.7 (C-4),163.0 (C-2),154.9 (C-8a),148.5 (C-5),146.4 (C-7),132.3 (C-1′),131.6 (C-4′),129.7 (C-6),129.4 (C-3′,5′),126.8 (C-2′,6′),104.9 (C-3),104.1 (C-5a),100.7 (C-8),54.4 (C-10,13),52.6 (C-9),27.6 (C-11,12);ESIMSm/z:386.106 8[M+H]+(calcd for C20H19NO5S,385.098 4)。
2.2 黄芩素酯类衍生物的制备
参考文献[13],称取相应有机酸2.2 mmol,加入30 mL的无水四氢呋喃,在-5 ℃条件下搅拌,然后称取2.6 mmol的N-羟基苯并三氮唑(HOBT),2.6 mmol的二环己基碳酰二亚胺(DCC),溶于20 mL无水四氢呋喃,然后用滴液漏斗将其滴加到上述反应液中于-5 ℃条件下搅拌制备活性酯,12 h后将反应液过滤,滤液备用。
称取1 g化合物1(2.6 mmol),加入50 mL无水四氢呋喃,在搅拌条件下,依次加入0.43 mmoL二甲氨基吡啶(DMAP)、0.8 mL三乙胺,在室温下搅拌,然后将上述活性酯滤液用滴液漏斗将其缓慢滴加到此反应液中继续反应,薄层色谱法监测反应进程,待反应达到平衡。将反应液浓缩至干,用硅胶柱层析分离(洗脱剂为二氯甲烷/甲醇),必要时进一步重结晶得目标化合物。
2.2.1 5,6-二羟基-4-氧代-2-苯基-8-(硫代吗啉亚甲基)-4H-苯并吡喃-7-基苯甲酸酯(化合物2)的制备
称取苯甲酸0.268 g,按通法制备,得产物0.89 g,黄色晶体,产率70.1%;1H NMR (DMSO-d6,600 MHz)δ:13.14 (s,1H,5-OH),8.12~8.14 (m,4H,H-2′,6′,2′,6′),7.76 (t,J=7.5 Hz,1H,H-4′),7.60-7.64 (m,5H,H-3′,4′,5′,3′,5′),7.01 (s,1H,H-3),4.20 (s,2H,H-9),3.06(s,4H,H-11,14),2.75 (s,4H,H-12,13);13C NMR (DMSO-d6,150 MHz)δ:181.7 (C-4),163.5 (C-10),162.6 (C-2),160.6 (C-8a),152.3 (C-5),151.5(C-7),133.8(C-4′),131.9(C-4′),130.8 (C-1′),129.7 (C-2′,6′),129.1 (C-3′,5′),128.8 (C-3′,5′),128.6 (C-1′),126.4 (C-2′,6′),122.5 (C-6),104.7 (C-3),102.0 (C-5a),98.8 (C-8),54.8 (C-11,14),53.3 (C-9),26.2 (C-12,13);ESIMSm/z:490.134 6[M+H]+(calcd for C27H23NO6S,489.124 6)。
2.2.2 5,6-二羟基-4-氧代-2-苯基-8-(硫代吗啉亚甲基)-4H-苯并吡喃-7-基-4-甲氧基苯甲酸酯(化合物3)的制备
称取对甲氧基苯甲酸0.334 g,按通法制备,得产物0.92 g,淡黄色粉末,产率68.3%;1H NMR (DMSO-d6,600 MHz)δ:13.09 (s,1H,5-OH),8.12 (dd,J=6.8 Hz,1.6Hz,2H,2H-2′,6′),8.04 (d,J= 8.8 Hz,H-2′,6′),7.60-7.65 (m,3H,H-3′,4′,5′),7.06 (d,J=8.8 Hz,2H-3′,5′),7.02 (s,1H,H-3),4.19 (s,2H,H-9),3.84 (s,3H,H-15),3.08 (s,4H,H-11,14),2.74 (s,4H,H-12,13);13C NMR (DMSO-d6,150 MHz)δ:182.2 (C-4),164.0 (C-10),163.7 (C-4′),163.0 (C-2),161.4 (C-8a),152.8 (C-5),152.2 (C-7),132.4 (C-2′,6′,4′),131.4 (C-1′),129.7 (C-3′,5′),126.9 (C-2′,6′),123.3(C-6),121.3 (C-1′),114.6 (C-3′,5′),105.2 (C-3),102.6 (C-5a),99.4 (C-8),56.1 (C-15),53.9(C-11,14),53.3 (C-9),26.7 (C-12,13);ESIMSm/z:520.144 0[M+H]+(calcd for C28H25NO7S,519.135 2)。
2.2.3 5,6-二羟基-4-氧代-2-苯基-8-(硫代吗啉亚甲基)-4H-苯并吡喃-7-基-2-溴苯甲酸酯(化合物4)的制备
称取邻溴苯甲酸0.439 g,按通法制备,得产物0.98 g,淡黄色粉末,产率66.5%;1H NMR (DMSO-d6,600 MHz)δ:13.18 (s,1H,5-OH),8.12-8.15(m,3H,H-2′,6′,6′),7.83~7.86 (m,1H,H-4′),7.57~7.64 (m,5H,H-3′,4′,5′,3′,5′),7.00 (s,1H,H-3),4.23 (s,2H,H-9),3.11 (s,4H,H-11,14),2.78 (s,4H,H-12,13);13C NMR (DMSO-d6,150 MHz)δ:182.1 (C-4),163.1 (C-10),163.0 (C-2),161.6 (C-8a),153.1 (C-5),152.2 (C-7),135.0 (C-4′),134.5 (C-1′),132.6 (C-3′),132.4 (C-6′),131.4 (C-4′),130.7 (C-1′),129.7 (C-3′,5′),128.4 (C-6),126.9 (C-2′,6′),123.1 (C-5′),121.9 (C-2′),105.2 (C-3),102.3 (C-5a),99.1 (C-8),53.8 (C-11,14),53.2 (C-9),26.6 (C-12,13);ESIMSm/z:568.031 5[M+H]+(calcd for C27H22BrNO6S,567.035 1)。
2.2.4 5,6-二羟基-4-氧代-2-苯基-8-(硫代吗啉亚甲基)-4H-苯并吡喃-7-基甲基丙烯酸酯(化合物5)的制备
量取α-甲基丙烯酸0.189 mL,按通法制备,得产物0.74 g,淡黄色晶体,产率59.8%;1H NMR (DMSO-d6,600 MHz)δ:13.11 (s,1H,5-OH),8.11 (dd,J=6.8 Hz,1.6 Hz,2H,H-2′,6′),7.58~7.64 (m,3H,H-3′,4′,5′),6.99 (s,1H,H-3),6.23 (s,1H,H-12E),5.89 (s,1H,H-12Z),4.16 (s,2H,H-9),3.03 (s,4H,H-14,17),2.76 (s,4H,H-15,16),2.01 (s,3H,H-13);13C NMR (DMSO-d6,150 MHz)δ:182.3 (C-4),164.7 (C-10),163.2 (C-2),160.6 (C-8a),152.7 (C-5),152.0 (C-7),135.4 (C-11),132.5 (C-4′),131.3 (C-1′),129.7 (C-3′,5′),128.2 (C-6),126.9 (C-2′,6′),123.0 (C-12),105.2 (C-3),102.7 (C-5a),99.4 (C-8),53.9 (C-14,17),53.3 (C-9),26.8 (C-15,16),18.6 (C-13);ESIMSm/z:386.106 8[(M-acid)+H]+(calcd for C24H23NO6S,453.124 6)。
2.2.5 5,6-二羟基-4-氧代-2-苯基-8-(硫代吗啉亚甲基)-4H-苯并吡喃-7-基苯丙烯酸酯(化合物6)的制备
称取肉桂酸0.325 g,按通法制备,得产物0.93 g,淡黄色晶体,产率69.5%;1H NMR (DMSO-d6,600 MHz)δ:13.11 (s,1H,5-OH),8.11 (dd,J=6.8 Hz,1.6 Hz,2H,H-2′,6′),7.85 (d,J= 16.1 Hz,H-16),7.80~7.82 (m,2H-2′,6′),7.59~7.65 (m,3H,H-3′,4′,5′),7.45-7.48 (m,3H,H-3′,4′,5′),7.01 (s,1H,H-3),6.91 (d,J=16.1 Hz,1H,H-15),4.18 (s,2H,H-9),3.04 (s,4H,H-11,14),2.76 (s,4H,H-12,13);13C NMR (DMSO-d6,150 MHz)δ:182.3 (C-4),164.4 (C-10),163.2 (C-2),160.6 (C-8a),152.7 (C-5),152.1 (C-7),146.8 (C-16),134.4 (C-1′),132.5 (C-4′),131.3 (C-1′),131.3(C-1′),129.7 (C-3′,5′),129.5(C-3′,5′),129.1 (C-2′,6′),126.9 (C-2′,6′),123.0 (C-6) ,117.4 (C-15),105.2 (C-3),102.8 (C-5a),99.4 (C-8),53.9 (C-11,14),53.3 (C-9),26.8 (C-12,13);ESIMSm/z:516.150 5[M+H]+(calcd for C29H25NO6S,515.140 3)。
2.3 目标化合物的结构确证
通过1H NMR、13C NMR数据以及质谱的分子离子峰与目标化合物的相对分子质量比较,确认了通过酯化反应所合成的目标化合物为单取代。由于5位酚羟基与4位羰基形成氢键,反应活性变钝,不易发生酯化反应,且氢谱中化学位移12.9左右的单峰存在,可以确认分子中有5位酚羟基存在,即5位没有被取代。然而由于分子结构中6,7位酚羟基上的氢为活泼氢,在核磁氢谱中没有出峰,因而不能确定6位和7位的取代基情况,通过氢谱、碳谱、质谱可得出的目标化合物的可能结构如图2。
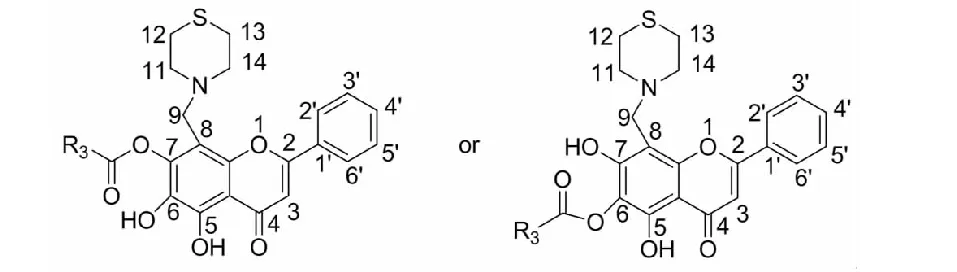
图2 目标化合物的推测结构Fig.2 The proposed structures of target compounds
由于酯键上没有氢原子,即使通过二维核磁HMBC谱,也不能确定其酰基化的位置是在6位还是在7位酚羟基上。本实验通过化学方法来确认其酰基化位置。由于分子中具有邻二酚羟基结构的黄酮类化合物在氨性甲醇溶液中,可与氯化锶(SrCl2)生成绿色至棕色乃至黑色沉淀[14],反应式如图3。通过该方法可以判定所合成的化合物分子中是否有邻二酚羟基,进而确定取代基的位置。其确定依据为:若是黄芩素6位酚羟基没有被取代成酯,化合物6位与5位酚羟基形成邻二酚羟基,用该法将成阳性反应,则可以说明化合物的酯键是在7位。若是黄芩素6位酚羟基成酯被取代,化合物分子中无邻二酚羟基,用该法将成阴性反应,则可以证明化合物的酯键是在6位。

图3 含邻二酚羟基黄酮类化合物与氯化锶的反应Fig.3 The reaction of O-diphenol hydroxy compounds with ruthenium chloride
实验方法:取约1.0 mg检品置小试管中,加入1.0 ml甲醇溶解(必要时可在水浴上加热),加入3滴0.01 mol/L氯化锶的甲醇溶液,再加3滴用氨蒸气饱和的甲醇溶液,注意观察有无颜色变化和沉淀生成[14]。
本实验选择结构已知的化合物:黄芩素,白杨素,黄芩苷,金合欢素,8-硫代吗啉亚甲基黄芩素,以及本实验室合成并且通过氢谱,碳谱,质谱以及二维谱HMBC结构确定的7-苄基黄芩素作为对照,和上述合成的目标化合物进行SrCl2反应实验。对照品结构如图4。
实验结果显示黄芩素,黄芩苷,7-苄基黄芩素,8-硫代吗啉亚甲基黄芩素,具有邻二酚羟基的化合物成阳性反应。白杨素,金合欢素,无邻二酚羟基的化合物呈阴性反应。本实验所合成的化合物均成阳性反应。因此从实验结果可知,本实验所合成的化合物分子中具有邻二酚羟基,6位与5位酚羟基形成邻二酚羟基,即6位没有取代基。则证明化合物分子中的酰基接到了7位。综合波谱数据和化学的方法可以确定本实验所合成的化合物为,7位酚羟基成酯,8位连接胺亚甲基的系列化合物,其结构如图5。
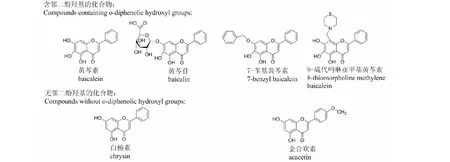
图4 对照化合物的结构Fig.4 The structures of control compounds

图5 目标化合物的结构Fig.5 The structures of target compounds
2.4 目标化合物抗MCF-7肿瘤细胞的活性评价
2.4.1 待测化合物的配制
将待测化合物和黄芩素溶解于DMSO中,配制成5 mg/mL的母液,用0.2 μm过滤器过滤母液,用饥饿培养基将化合物稀释成50、10、2、0.4、0.08 mg/mL的5个浓度的含药培养基,备用。
2.4.2 抗MCF-7肿瘤细胞活性测试
将MCF-7细胞(人乳腺癌细胞)稀释成每100 000/mL,取0.1 mL细胞悬液接种到96孔板,每孔细胞数量为10 000,培养24小时后,弃去旧培养基,换为饥饿培养基(1640培养基+2%FBS),培养12小时。弃去旧培养基,换为含药培养基,8个复孔每孔0.1 mL,培养72小时后,取出全部96孔板,每孔内加入CCK8试剂10 μL,37 ℃孵育1小时,在450 nm波长下,测量其OD值并打印数据,利用Graphpad Prism5分析数据,得到IC50值。
3 结果
本实验通过mannich反应,在黄芩素8位引入了硫代吗啉亚甲基,通过酯化反应在其7位引入不同的酯基,得到了6个目标化合物,其抗MCF-7肿瘤细胞活性结果如表1。

表1 目标化合物的抗肿瘤活性结果Table 1 Antitumor activity results of the target compounds
由活性结果可以看出,6个目标化合物中有4个化合物活性超过先导化合物黄芩素。在8位引入硫代吗啉胺亚甲基后(化合物1),活性比先导化合物强,在其7位再接入苯甲酸(化合物2)、α-甲基丙烯酸(化合物5)、肉桂酸(化合物6)成酯键后活性比先导化合物强,接入对甲氧基苯甲酸(化合物3)和邻溴苯甲酸(化合物4)后活性比先导化合物弱。
4 结论
本实验通过mannich反应和酯化反应,对黄芩素进行了结构修饰,在其8位引入了胺亚甲基,7位引入不同的酯基,得到了6个目标化合物,其中化合物2~化合物6为新化合物。由于黄芩素7-OH处于4位羰基的对位,在p-π共轭效应的影响下,反应活性较强。5位酚羟基与4位羰基形成氢键,活性变钝,难以发生反应,因此黄芩素酚羟基的活性是7位>6位>5位。但是在黄芩素8位接入胺亚甲基后,7位酚羟基有了一定的空间位阻,所以酯化反应的时候,酰基化反应发生在7位还是在6位,并不清楚。实验通过波谱手段对其结构进行了表征,并利用黄酮类化合物邻二酚羟基与金属盐类试剂的络合反应,巧妙而简单的确证了目标化合物的结构。同时确认了在文中的实验条件下,黄芩素8位接入胺亚甲基后,酯化反应不受空间位阻的影响,仍能发生在活性较强的7位酚羟基上。通过抗MCF-7肿瘤细胞活性实验可以看出目标化合物在8位引入含氮原子的胺亚甲基后活性比先导化合物黄芩素强,在其7位上再引入酯键后有的化合物活性比先导化合物强,有的则比先导化合物活性差。在其7位上再引入苯甲酸、α-甲基丙烯酸、肉桂酸成酯键后活性比黄芩素强,引入对甲氧基苯甲酸和邻溴苯甲酸后活性比先导化合物弱。分析原因可能是由于在苯甲酸的邻位和对位有较大的基团存在,不利于化合物与靶点的结合,使得活性比黄芩素差。
