文冠果果实转录组测序及分析
2019-07-26赵阳阳郭雨潇张凌云
赵阳阳 郭雨潇 张凌云
(北京林业大学 森林培育与保护教育部重点实验室,北京 100083)
文冠果(Xanthoceras sorbifoliaBunge)又名文冠花、文光花、僧灯毛道,隶属无患子科(Sapindaceae)文冠果属(XanthocerasBunge),单属单种。一般为亚乔木或大灌木,遍布华北、华东及西北地区[1-2]。文冠果具有较强的适应性和抗逆能力,种子营养丰富、种仁含油率高,是一种珍贵的木本油料植物,有北方油茶之称,具有巨大的发展潜力[3]。目前对于文冠果的研究多集中在生长发育[4-6]、栽培管理[7-8]、油脂组成及提取[3,5,9-11]、生物柴油制备[12-13]和常规育种[14]等方面,也有一些基因克隆和功能分析[15-19],关于果实生长发育过程中的分子机理研究很少。
转录组测序研究能够从整体水平研究基因功能与基因结构并揭示特定生物学过程的分子机理,广泛应用于分析生物和医学研究等领域[20]。近年来,随着高通量测序技术发展,测序时间和成本显著降低,很多模式和非模式植物完成了转录组测序,文冠果的转录组测序也取得了很大进展。Liu 等[21]对文冠果芽、花、叶和种子的混合样进行高通量转录组测序,构建了文冠果参考转录组。敖妍等[22]应用Illumina Hi-seqTM2000高通量测序技术对文冠果花芽进行了转录组分析来解析文冠果花芽形态分化的分子调控模式与机制。刘玉林等[23]在对7个省份16份文冠果叶片进行转录组测序的基础上分析和筛选了转录组序列中的SSR标记,并设计开发ESTSSR标记,为文冠果的遗传多样性分析提供标记资源。申展等[24]运用SSR标记的方法,研究了全国14个地区具有代表性的138份文冠果种质资源的遗传多样性,并逐步构建出25份文冠果核心种质。文冠果果实为主要收获部位,果实发育和营养物质积累决定了产量和经济效益,是文冠果研究的重点,而文冠果果实发育过程分子机理尚不清楚。
本研究利用Illumina Hi-seqTM2000高通量测序平台对两个发育时期的文冠果果实进行转录组测序,可以全面了解文冠果果实基因表达情况,获得大量基因序列,同时可以开发SSR分子标记,为后期文冠果品种鉴定和分子育种提供依据。
1 材料与方法
1.1 材料
文冠果果实采集于北京市圆明园内长势良好,无病虫害的20年生实生树。通过物候观察,样品采集时间分别为2017年盛花后20 d(1号样)和60 d(2号样),分别在树体东南西北不同方位各采集3个果实,混匀,然后立即放入液氮中带回实验室,-80℃保存备用。
1.2 方法
1.2.1 RNA提取与文库构建 两个时期样品均混合12个果实提取RNA来建库测序,以消除个体误差。总RNA提取采用植物总RNA提取试剂盒(Omega,美国),NanoDropTM2000 分光光度计(Thermo Fisher,美国)和琼脂糖凝胶电泳检测RNA的纯度、浓度和完整性。检验合格的文冠果果实总RNA,用带有Oligo(dT)的磁珠富集mRNA,加入Fragmentation Buffer 使其成为短片段,用六碱基随机引物(random hexamers)合成cDNA第1链,然后加入缓冲液、dNTAs、RNase H和DNA polymerase I合成cDNA第二链;再经过QiuQuick PCR试剂盒(QIAGEN,德国)纯化并加 EB 缓冲液洗脱之后做末端修复、加poly(A)并加测序接头,再经琼脂糖凝胶电泳回收目的片段,最后进行PCR扩增构建测序文库。
1.2.2 转录组测序与组装 利用Illumina HiseqTM2000对文冠果果实测序文库进行高通量测序。测序仪产生的原始图像数据经 base calling 转化为序列数据raw reads,经过平台过滤除杂和冗余去除后得到高质量的clean reads,然后使用短reads 组装软件Trinity做转录组de novo组装获得Unigene。
1.2.3 Unigene功能注释 使用BLAST 软件将Unigene序列与NR(非冗余蛋白数据库)、Swiss-Prot(蛋白质序列数据库)、GO(基因本体论数据库)、COG/KOG(蛋白质原核/真核同源数据库)、eggNOG(直系同源蛋白数据库)、KEGG(京都基因与基因组百科全书)数据库对比,使用KOBAS2.0得到Unigene在KEGG中的KEGG Orthology结果,预测完Unigene的氨基酸序列之后使用HMMER软件与Pfam(蛋白质家族域数据库)数据库对比,获得Unigene的注释信息。
1.2.4 SSRs(Simple Sequence Repeats) 检 索 与 分析 利用MISA软件检测文冠果果实转录组Unigene,搜索其中的SSRs并进行统计分析。
2 结果
2.1 测序结果及从头组装
碱基质量值(Quality score)是碱基识别(Base calling)出错的概率的整体映射。碱基质量值越高表面碱基识别越可靠。文冠果果实发育早期样本测得的Q30碱为89.51%,测得序列的GC含量占44.62%;发育后期样本测得的Q30碱基百分比为89.60%,测得序列的GC含量占44.61%。经过测序质量控制,共得到42 545 764个序列读取片段(Reads):其中包含了12.74 Gb核苷酸序列信息,各样品Q30碱基百分比均不小于89.51%,说明转录组测序质量较高,可以进行下一步的数据组装。
测序数据组装共获得157 244个转录本,序列信息达261 799 238 bp,N50为2 693 bp,平均长度为1 664.92 bp。所得转录本序列进一步组装得到68 298个单基因簇(Unigene),序列信息总长度为59 281 489 bp,N50为 1 589 bp, 平 均 长 度 为867.98 bp。其中长度在300-500 bp的Unigene占比最高,为21.15%,长度在1 kb以上的Unigene占比24.49%(表1)。组装完整性较高,可用于后续的注释分析。

表1 文冠果果实转录组数据组装结果
2.2 功能基因注释
利用BLAST软件将获得的Unigene数据信息分别在 Nr、Swiss-Prot、Pfam、GO、KOG和 KEGG数据库比对,对所有注释上的Unigene数目进行分析统计。结果(表2)表明文冠果果实两个样本共包含68 298个Unigene,其中24 691个Unigene得到注释,43 607个Unigene未得到注释,获得注释的Unigene占总Unigene的36.15%。在注释的Unigene中,Nr数据库注释的Unigene最多,达到24 059个;COG数据库注释的Unigene最少,仅有7 483个。注释到长度300≤ 长度< 1 000 Unigene数为7 724个,其中注释到Nr数据库300≤长度< 1 000的最多,达到7 425个;注释到COG数据库300≤长度< 1 000 Unigene数最少,有1 544个。注释到长度≥ 1 000个碱基的Unigene数为13 924个,其中注释到Nr数据库长度≥ 1 000 Unigene数最多,达到13 892个。
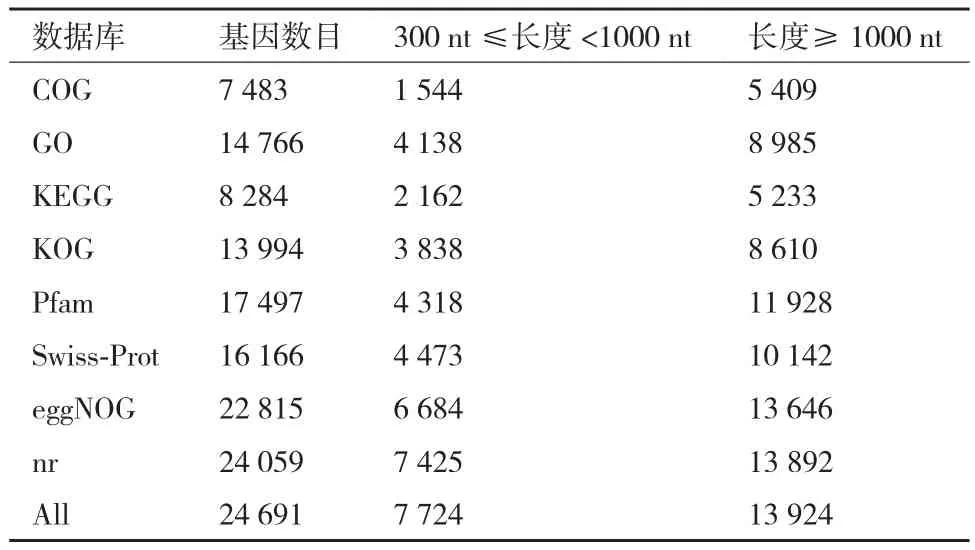
表2 文冠果果实基因的BLAST比对结果
根据Nr数据库注释显示,Unigene注释同源基因的物种分布图如图1所示,甜橙Citrus sinensis的同源序列最多,达7 131条,占注释序列总数的29.65%,其次是克莱门柚Citrus clementina,匹配同源序列4 950条,占总注释序列的20.58%,两个物种所匹配的同源序列占总注释序列的一半。其次匹配较高的分别是可可Theobroma cacao(1 916条,7.97%)、 葡 萄Vitis vinifera(1 539条,6.40%)、稻Oryza sativa(700条,2.91%)、毛果杨Populus trichocarpa(686,2.85%)、麻风树Jatropha curcas(662,2.75%)、蓖麻Ricinus communis(595,2.47%)、胡杨Populus euphratica(457,1.90%)、桃Prunus persica(377,1.57%)和其他物种(5 039,20.95%)。

图1 文冠果果实转录组Unigene与Nr数据库匹配物种分布
为了进一步揭示文冠果果实转录组Unigene的功能分类,根据Nr数据库注释得到的信息,通过GO数据库,对文冠果果实转录组Unigene进行基因生物学特征功能分类。结果(图2)显示,GO数据库注释的14 766个文冠果果实转录组Unigene可分为细胞组分(Cellular component)、分子功能(Molecular function)和生物过程(Biological process)等3个GO类别的54个小组。细胞组分涉及28 681个GO条目,分为17个功能组,其中细胞(6 588个)、细胞部分(6 588个)和细胞器(5 044个)涉及的Unigene较多;分子功能涉及18 283个GO条目,分为17个功能组,其中催化活性(7 790个)和结合活性(7 637个)含Unigene较多;41 373个GO条目归属于生物过程,分为20个功能组,其中代谢过程(10 120个)、细胞进程(8 512个)和单一生物体过程(7 194个)涉及的Unigene较多。
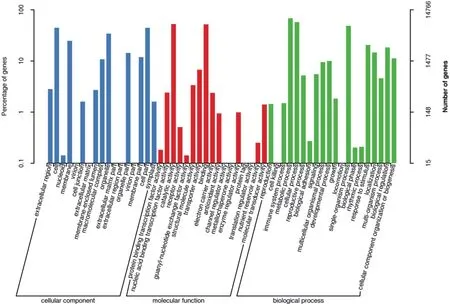
图2 文冠果果实转录组Unigene的GO功能分类图
为了进一步分析文冠果果实转录组Unigene的功能,进行了KOG功能分类分析,结果如图3所示,共获得25个不同的功能分类,比较全面,包含了大多数的生命活动。其中,一般功能预测(General function prediction only)的基因数量最多3 998条(25.32%),是最大的功能类群,其次是翻译后修饰、蛋白质翻转和分子伴侣(Posttranslational modification,protein turnover,chaperones)1 387条(8.62%),信号转导类机制(Signal transduction mechanisms) 次 之, 由 1 254条(8.17%), 碳 水化合物转运与代谢(Carbohydrate transport and metabolism)也有870条(5.41%),另外划分到脂类转运与代谢(Lipid transport and metabolism)这一功能类别的有560条(3.61%),其他功能分类的基因数量不尽相同。
以KEGG数据库为依据,将8 284个文冠果果实转录组Unigene分为127个代谢通路(表3)。核糖体(445个)、碳代谢(320个)、氨基酸的生物合成(288个)、内质网上蛋白质处理(240个)和植物激素信号转导(221个)等代谢通路涉及的Unigene较多。此外,淀粉和糖代谢通路涉及200个Unigene,脂肪酸代谢通路有81个,脂肪酸生物合成、不饱和脂肪酸的生物合成和脂肪酸延长分别涉及40、40和30个Unigene。

图3 文冠果果实转录组Unigene的KOG注释分析
2.3 转录组SSRs特征分布
利用MISA软件对筛选得到的1 kb以上的Unigene做SSR分析,结果如表4所示:共发现SSR位点11 732个。其中,单核苷酸SSR最丰富,有7 139个,远远超过其他种类SSR,占全部SSR的60.85%,平均长度最短,为12.46 bp;二核苷酸SSR(2 655个)和三核苷酸SSR(1 854个)分别占总SSR的22.63%和15.8%,平均长度分别为15.47 bp和16.53 bp;四核苷酸SSR(63)、五核苷酸SSR(11)和六核苷酸SSR(10)很少,三者之和不到总SSR的1%,其平均长度较长,六核苷酸SSR的平均长度达到43.2 bp。
3 讨论
文冠果适应性广,抗逆性强,含油率高,是我国重点发展的木本油料树种,具有巨大的发展潜力[25-27]。目前关于文冠果的研究主要集中在生长发育[4-6]、栽培管理[7-8]、油脂组成及提取[3,5,9-11]、生物柴油制备[12-13]和常规育种[14]等方面,分子生物学方面的研究较少,果实发育过程中的基因表达情况尚不清楚。第二代高通量测序技术因测序时间短、成本低和数据量大的优点近年来被广泛应用于组学研究中。本研究利用高通量测序技术得到了文冠果果实的转录组数据,共获得12.74Gb Clean Data,各样品Clean Data均达到6.31Gb,Q30碱基百分比在89.51%及以上,测序质量较高。组装后获得了68 298个Unigene序列,其中长度在1 kb以上的Unigene有16 724条,组装完整性良好。
Nr数据库注释结果显示,由于缺少文冠果基因组和转录组,大量Unigene未获得注释,其中仅有24 691个Unigene得到注释,占36.15%。这与松萝凤梨[28]、白鲜根[29]等已报道物种转录组测序注释结果相似,说明文冠果果实转录组有大量序列需要深入挖掘。与甜橙Citrus sinensis的同源序列最多,达7 131条,占注释序列总数的29.65%,其次是克莱门柚Citrus clementina,匹配同源序列4950条,占总注释序列的20.58%,两个物种所匹配的同源序列占总注释序列的一半。

表3 文冠果果实转录组Unigene的KEGG注释
通过GO数据库对文冠果果实转录组Unigene进行基因生物学特征功能分类,代谢过程(10 120个)、细胞进程(8 512个)、催化活性(7 790个)和结合活性(7 637个)等含Unigene较多,总体来说,文冠果果实在细胞活动、代谢活动的基因表达丰度较高,表明文冠果果实自身具有较强的代谢能力。
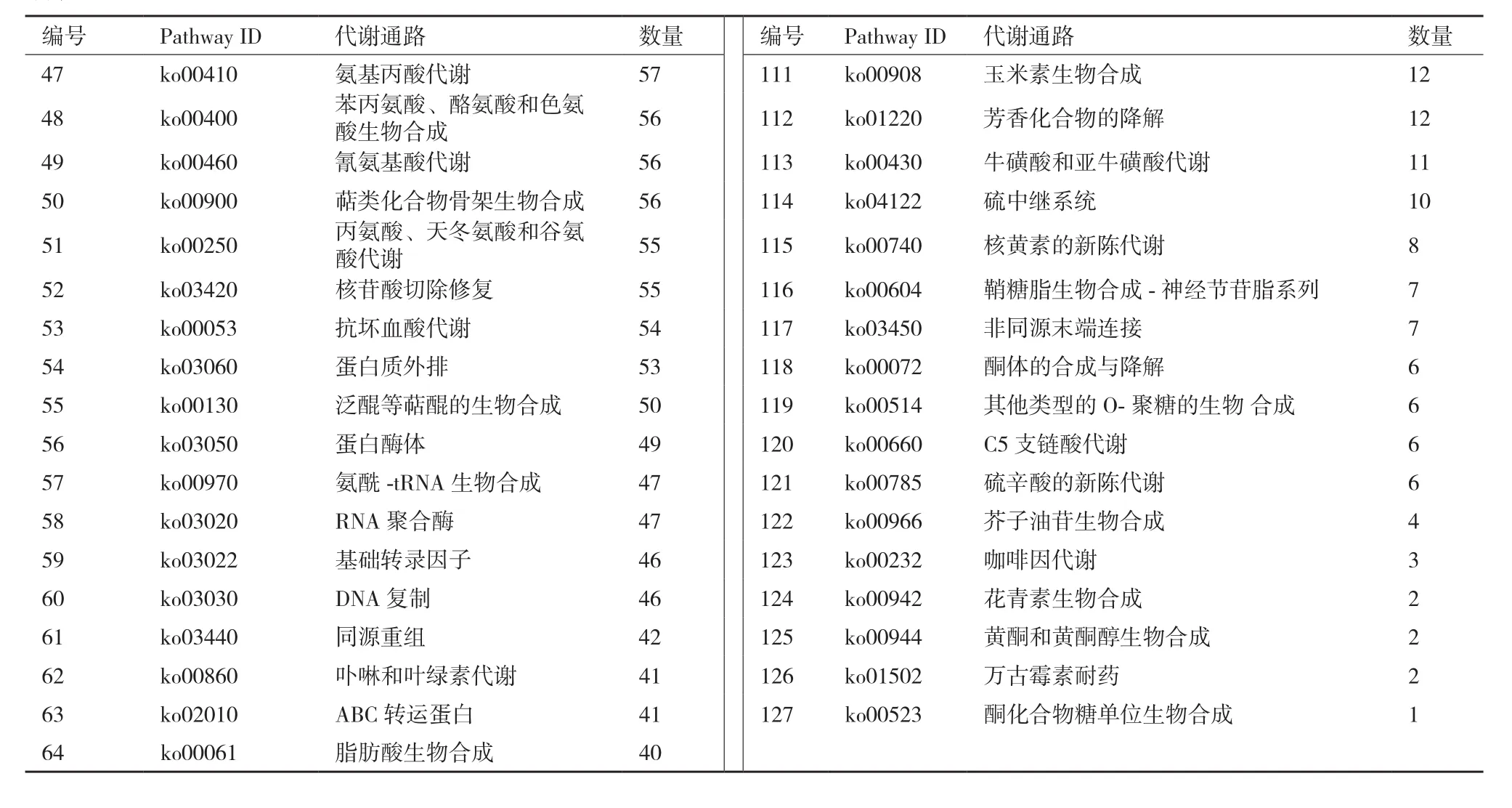
续表

表4 文冠果果实转录组SSR分布特征
通过KOG数据库注释共获得25个不同的功能分类,比较全面,包含了大多数的生命活动,文冠果果实转录组中Unigene主要涉及基因表达、蛋白质合成和物质代谢等功能分类,符合文冠果果实作为强“库”器官积累营养物质这一基本功能。
以KEGG数据库为依据,将8 284个文冠果果实转录组Unigene分为127个代谢通路,与Liu等[21]和敖妍等[22]研究结果不同,推测可能是由于取材不同造成的。本研究样品为文冠果果实,而Liu等测序样品为文冠果芽、花、叶和种子的混合样,敖妍等以文冠果花芽为材料。本研究分离了大量涉及淀粉和糖代谢、脂肪酸代谢、脂肪酸生物合成、不饱和脂肪酸的生物合成和脂肪酸延长等通路相关基因,为后期研究文冠果果实生长发育和营养物质积累奠定基础。
目前限制文冠果产业发展的一大因素就是种质资源混乱,缺少优良品种,仅利用外部形态学特征很难做到精细区分。转录组数据除了用于基因挖掘及表达调控研究外,可以开发大量EST-SSR分子标记。SSR标记是近年来发展起来的一种以特异引物PCR为基础的分子标记技术,广泛用于遗传图谱的构建、目标基因的标定、指纹图谱的绘制等研究中[20]。利用MISA软件对筛选得到的1kb以上的Unigene做SSR分析,共发现SSR位点11 732个,远远高于刘玉林等[23]和申展等[24]研究结果中的SSR位点,分析是取材不同造成的结果差异。后期会将实验结果与之进行比对,筛选验证开发出更可靠的SSR位点,用于文冠果种质资源分类和优良品种培育。
4 结论
本研究利用Illumina Hi-seqTM2000高通量测序平台对两个发育时期的文冠果果实进行转录组测序,共获得68 298个Unigene序列,共有24 691个Unigene得到注释,占36.15%;GO数据库注释显示14 766条Unigene分为细胞组分、分子功能及生物学过程3大类54个功能组;KOG数据库中注释到的13 994条Unigene功能系统分为25类;以KEGG代谢途径数据库为依据,可将8 284个文冠果果实转录组Unigene分为127个代谢通路,可以全面了解文冠果果实的代谢途径信息;在文冠果果实转录组中发现11 732个SSR位点,最多的为单核苷酸SSR,占60.85%。
