基于CRISPR/Cas9加工番茄α-Man突变体的构建
2019-07-26张圆圆邵冬南崔百明
张圆圆 邵冬南 崔百明
(石河子大学生命科学学院 石河子大学农业生物技术重点实验室,石河子 832003)
番茄(Solanum lycopersicum)属于呼吸跃变型果实,在成熟过程中乙烯产量骤增,果实迅速软化,影响商业价值。因此,探索能够抑制番茄果实过度软化、延长货架期、便利储存运输、增加销售品质的方法显得尤为重要[1-3]。Zhang等[4]研究表明果实的软化主要由于整个成熟过程中果实细胞壁组分和结构的破坏,但确切机制仍不清楚。Roberts等[5]在植物细胞壁以及其他细胞器中均发现有糖基化的N-糖蛋白存在。并且在果实成熟前期,用衣霉素(蛋白质糖基化抑制剂)处理后,番茄红素和类胡萝卜素等相关营养成分的积累、叶绿素降解以及果皮组织产乙烯能力都受到抑制,这表明N-糖蛋白的糖基化在果实成熟过程中起重要作用[6]。游离N-聚糖作为糖基化的前体或N-糖蛋白水解产物,在果实成熟阶段果皮组织中含量明显增加[7]。糖苷水解酶(Glycoside hydrolase)作用于N-糖蛋白的各种糖苷或寡糖,使糖苷键水解形成游离N-glycans,而缺乏寡糖部分的蛋白质会直接影响果实细胞壁与细胞膜的化学组分,从而破坏细胞的完整性,导致果实软化[8-9]。α-甘露糖苷酶(α-Man)切割糖蛋白中存在的高甘露糖型和植物复合型N-聚糖的末端α-甘露糖苷键,番茄α-Man在果实成熟时诱导表达,调节果实成熟软化过程。过表达α-Man导致番茄果实过度软化,而α-Man表达受抑制的RNAi植株,果实软化速率下降,硬度增加,保质期延长[10]。
目前,对于降低番茄果实软化程度的问题,通过RNAi抑制细胞壁修饰相关基因是最常用的方法,但果实风味严重受损[11-13]。Meli等[10]证明α-Man的RNAi植株果实成熟延缓,但营养成分不发生改变。基因组靶向修饰已成为改造基因组和研究基因功能的一个重要手段[14]。而基于CRISPR/Cas9系统的基因组编辑是使用最广泛的基因编辑技术,它由Cas9核酸酶和靶向基因组序列的指导RNA(gRNA)组成,能够简单、精准、高效地编辑单个基因的一个或多个靶位点,甚至多个基因的多个位点[15]。已成功应用于多种模式植物和作物[16-18]。
本研究通过CRISPR/Cas9基因组编辑手段,靶向加工番茄N-糖蛋白水解关键酶基因α-Man。通过农杆菌介导的遗传转化获得α-Man突变体植株,从而达到抑制N-糖蛋白降解、降低加工番茄成熟期果实软化程度的目的,具有重要的生物学意义及商业价值。
1 材料与方法
1.1 材料
加工番茄(Solanum lycopersicum)品种里格尔(87-5)作野生型;由上海生工生物工程股份有限公司合成sgRNA表达框并完成测序工作。
1.2 方法
1.2.1 植物表达载体构建α-Man敲除靶点引物序列设计:首先根据加工番茄α-Man序列(EU244853),利用在线软件CRISPR-PLANT(https://www.genome.arizona.edu/crispr/CRISPRsearch.html)筛选合适的编辑靶点。然后根据PAM(NGG)位点切割原则设计靶序列sg_Man_1S,并给出它的互补序列sg_Man_1A(表 1)。

表1 引物序列列表
载体构建:首先以hcas9-S/hcas9-A(表1)为引物扩增hCas9目的片段,并通过NcoⅠ/BstEⅡ克隆到pCAMBIA1302载体上,构建p1302-Cas9载体;用XhoⅠ酶切载体pBP-35S和p1302-Cas9,以Kan抗性基因替换载体骨架p1302-Cas9的Hyg抗性基因,构建pKAN-Cas9载体;然后将公司合成的sgRNA表达框(图1)克隆到pUC19载体上,构建pUC-sgRNA载体;Hind Ⅲ/EcoRⅠ双酶切载体pUC-sgRNA和pKAN-Cas9,构建pKAN-sgR-Cas9载体;最后通过引物退火的方式将sg_Man_1S/sg_Man_1A结合成带黏性末端的双链DNA片段,并利用Ⅱs型限制酶AarⅠ克隆到pKAN-sgR-Cas9载体上,构建植物表达载体pKAN-sgR-Cas9-Man(图2)。

图1 sgRNA表达框示意图
测序正确的质粒电击转化根瘤农杆菌GV3101,通过菌落PCR筛选阳性克隆,活化单班用于番茄外植体侵染。

图2 植物表达载体pKAN-sgR-Cas9-Man结构示意图
1.2.2 加工番茄遗传转化 首先用2%次氯酸钠溶液浸泡消毒(摇床内,120 r/min,10 min)番茄种子,并用无菌水漂洗8-10次,播种于1/2MS(MS盐2.315 g/L+蔗糖15g/L+琼脂7.0 g/L,pH5.8)培养基上。28℃暗培养2 d,待种子发芽后,28℃长日照(光照16 h/黑暗8 h)继续培养3-4 d;然后取子叶中段、叶柄和茎段,平铺在共培养基上(MS盐4.43 g/L+蔗糖30 g/L+植物凝胶1.5 g/L+IAA 0.1 mg/L+ZT 0.5 mg/L,pH5.8)培养,28℃长日照培养2 d;再用AIM合成培养基(含100 μmol/L乙酰丁香酮)制备侵染液(OD600=0.6-0.8),侵染外植体10-15 min后,将其转入表面铺有一层薄滤纸的共培养培养基,24℃暗培养2 d;之后将外植体转移至筛选培养基(MS盐4.43 g/L+蔗糖30 g/L+植物凝胶1.5 g/L+IAA 0.1 mg/L+ZT 0.5 mg/L+Carb 300 mg/L+Kan 50 mg/L,pH5.8),28℃长日照培养。每隔10-15 d将外植体转移至新的筛选培养基进行继代培养,继代培养3次。最后将分化再生得到的Kan抗性幼苗小心切下,并转移到生根培养基(1/2MS培养基+IAA 0.1 mg/L+Carb 300 mg/L+Kan 50 mg/L,pH5.8)中诱导生根。当番茄幼苗生长至成株(10 cm左右)后,将其顶端生长点或芽尖(包含生长节点)切下转移至扩繁培养基(1/2MS培养基+IAA 0.1 mg/L,pH5.8),待扩繁植株长出新叶,取少量叶片进行检测。
1.2.3 转基因植株分子鉴定 取1 g左右番茄叶片,SDS法提取番茄基因组DNA作模版,Colony-R和sg_Man_1A(表1)作引物,另加野生型番茄叶片基因组DNA作阴性对照,pKAN-sgR-Cas9-Man质粒(100×)作阳性对照,水作空白对照进行PCR鉴定。
1.2.4 番茄α-Man突变体检测 以番茄叶片基因组DNA为底物,特异性检测酶ScaⅠ进行酶切,回收纯化酶切产物,以纯化产物为模板,DT-Man-F和DT-Man-R(表1)为引物,野生型番茄叶片基因组DNA作阴性对照,PCR扩增加工番茄α-Man部分片段(包含靶序列)。扩增产物用琼脂糖凝胶纯化,进行TA克隆和菌落PCR鉴定,选择正确的单克隆,提取质粒继续进行酶切检测,选择鉴定正确的克隆测序。根据测序结果比对分析靶序列的编辑情况。
2 结果
2.1 CRISPR/Cas9基因编辑载体的构建
α-Man敲除靶点引物序列设计:α-甘露糖苷酶(α-Man)是植物体内N-聚糖加工的关键酶,参与N-聚糖形成和细胞壁合成。生物信息学分析表明番茄α-Man全长为11 001 bp,共含有30个外显子和29个内含子。为获得α-Man的移码突变体,在α-Man第1外显子上,起始密码子ATG后170 bp处设计长度为18 bp的靶位点序列:5'-GGTCGATCAGTACTATGTTGG-3',5'端是碱基G,与中间载体pKAN-sgR-Cas9经AarⅠ酶切后形成黏性互补末端,同时提高番茄U6启动子(SlU6)的转录效率。靶序列3'端为PAM序列TGG,PAM前5个碱基处设有ScaⅠ内切酶识别位点(图3),用于检测α-Man突变体。

图3 加工番茄α-Man靶序列设计示意图
SlU6驱动sgRNA的表达载体构建:植物表达载体pKAN-sgR-Cas9-Man以pCAMBIA1302为载体骨架,分别使用NcoⅠ/BstEⅡ位点和HindⅢ/EcoRⅠ位点将Cas9和sgRNA以5'-3'方向插入pCAMBIA1302载体中。并通过AarⅠ将18 bp靶序列融合到gRNA支架的5'末端,由SlU6驱动sgRNA表达框,花椰菜花叶病毒(CaMV)35S启动子驱动hCas9表达框。利用EcoRⅤ酶切鉴定表达载体,结果符合预期设计(图4-A),同时对阳性克隆进行测序验证,序列比对结果表明载体构建正确。

图4 靶向α-Man载体构建
2.2 加工番茄转基因阳性植株的获得
试验前期,试图通过瞬时转化的方法实现对加工番茄α-Man的编辑,但结果显示靶序列并没有发生突变。因此,选择农杆菌介导的遗传转化方式来获得加工番茄转基因植株。图4-B是质粒电击转化农杆菌菌落PCR鉴定结果,得到预期大小为374 bp的电泳条带,说明质粒转入农杆菌。
之后活化菌斑,经过加工番茄外植体制备、浸染、共培养和3次继代培养,约30-40 d得到再生植株,20 d后扩繁成株,同时取样提取基因组DNA。以Colony-R和sg_Man_1A为引物对上述样品进行分子鉴定,结果有14株扩增出374 bp大小的特异性条带,与阳性对照所得目的条带大小一致,而阴性对照和空白对照均没有扩增出条带,说明检测到14株转基因阳性植株(图5)。

图5 转基因番茄植株的PCR检测结果
2.3 α-Man突变体检测
将转基因植株每20-30 d扩繁一次,共扩繁3次,待扩繁茎段生长至成株,进行酶切和PCR检测,试验预期发生突变的样品可以扩增出704 bp的目的条带,而没有发生突变的样品将和野生型对照一样没有特征条带。检测结果显示有8个样品扩增出704 bp的目标条带(图6)。

图6 PCR扩增基因组DNA酶切产物
后期菌落PCR检测证明只有4号和7号2个样品可以扩增出目的条带,选择鉴定正确的14个单克隆进行测序,结果(图7)显示,在靶位点处有一个克隆发生52个碱基的缺失突变(克隆4-1,图7-C),而其余13个克隆均发生单碱基突变,包括C-T转换(克隆1-9,图7-A)和A-G转换(克隆4-1,图 7-B)。
2.4 转基因植株的表型观察
通过对转基因试管苗和α-Man突变体植株表型的观察和记录,发现与野生型加工番茄相比,它们的表型均无明显差异,而且检测到点突变和缺失突变的植株也没有明显的表型差异(图8)。这与α-Man属于果实成熟特异性表达蛋白的研究结果一致,在番茄生长发育各阶段检测不到α-Man酶活性,所以植株没有明显的性状。
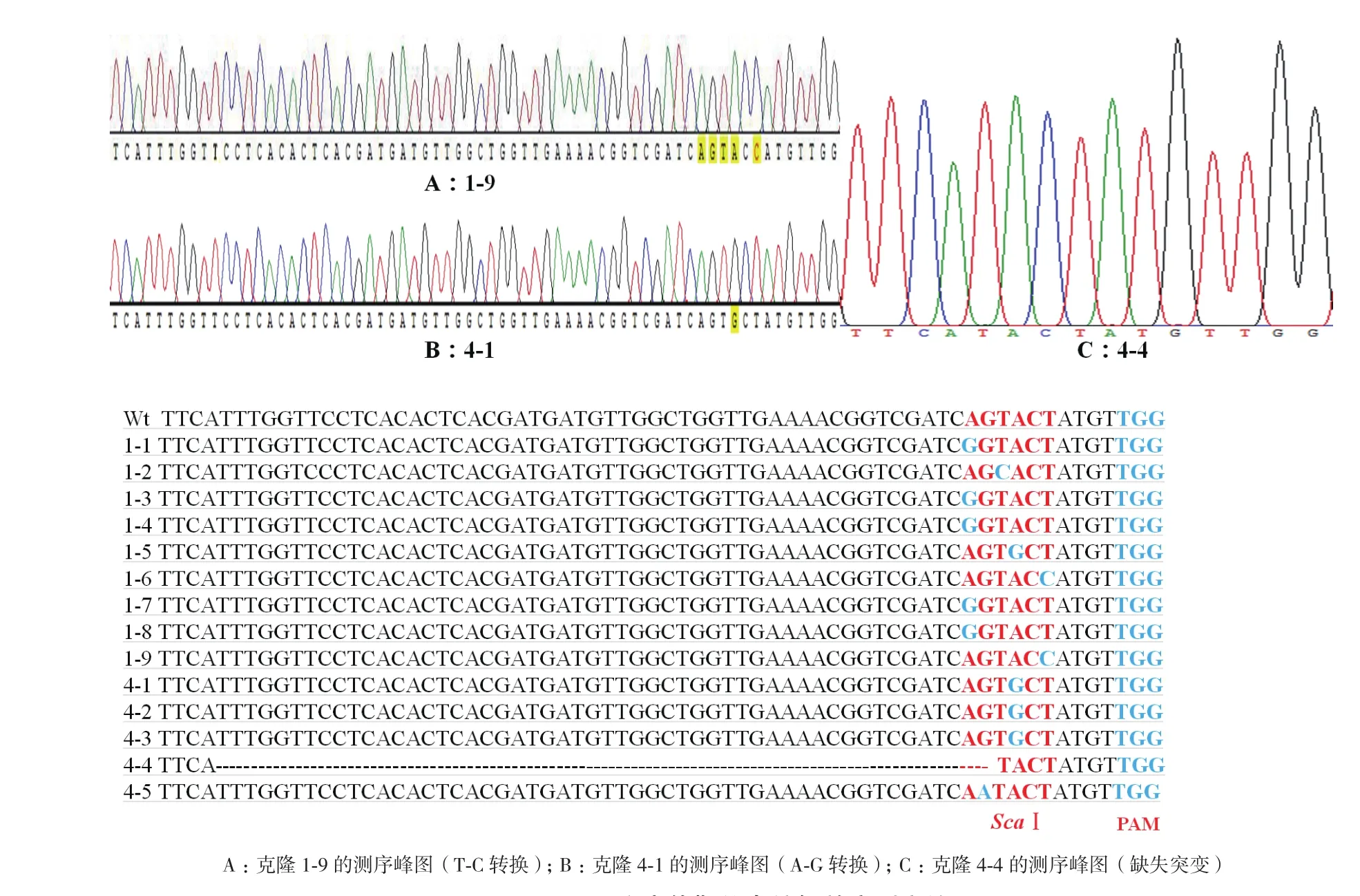
图7 α-Man突变体靶位点编辑效应测序结果

图8 转基因植株表型
3 讨论
细胞壁修饰相关蛋白α-Man主要参与蛋白质糖基化修饰、糖蛋白聚糖水解修饰和复杂N-聚糖的合成以及果实成熟软化,被认为是破坏细胞壁的关键因素,α-Man水解 α-1,2、α-1,3和 α-1,6糖苷键[10,19],促使 N-糖蛋白水解产生游离 N-glycans和缺乏寡糖部分的蛋白质,从而破坏细胞壁完整性,导致果实软化[8]。Meli等[10]研究表明α-Man在番茄果实成熟的破色期和粉红期阶段具有最大活性,而在植物的其他部分(例如茎、叶和根)中均未检测到α-Man活性。此外,番茄α-Man的RNAi植株果实硬度增加、货架期延长,并且与细胞壁降解和成熟相关基因的表达均下调。由此可见,通过抑制α-Man酶活性来降低番茄果实成熟软化是可行的。本研究利用CRISPR-Cas9系统靶向修饰加工番茄α-Man,在sgRNA的指导下,Cas9切割一条双链DNA链,产生平末端DNA双链断裂(DSB),再通过非同源末端连接(NHEJ)在DNA断裂位点处引入碱基置换、插入或缺失等突变[20]。目前,该技术已成功应用于多种植物体的瞬时或稳定表达,实例包括使用CRISPR/Cas9编辑系统产生的抗白粉病的小麦、耐受性的玉米、不完全成熟的番茄果实以及耐除草剂大豆等[21-24]。
试验由SlU6驱动sgRNA表达框,35S启动子驱动hCas9表达框,构建植物表达载体,在加工番茄中建立CRISPR/CAS9基因编辑体系。U6启动子是CRISPR/Cas9基因组编辑系统的重要元件之一,不同物种U6启动子驱动单个或多个sgRNA已经成功应用于CRISPR-Cas9系统中,但适用于番茄的U6启动子研究鲜有报道[25-27]。目前基于棉花 U6 启动子的CRISPR/Cas9基因编辑载体系统已在海岛棉中实现基因定点编辑[28],因此,本研究选择番茄U6启动子驱动sgRNA表达,结合hCas9蛋白编辑α-Man,成功在转基因加工番茄中检测到靶序列突变。TA克隆测序结果显示有2种编辑类型,一种表现为52 bp的缺失突变,对应的氨基酸序列改变,导致蛋白质翻译结果改变;另一种表现为单碱基突变,且突变位点多样,说明编辑效果不一致,效率较低,编辑类型较少,这与棉花原生质体和大豆根尖的基因编辑结果类似,Chen等[29-30]瞬时转化棉花原生质体后检测突变结果多为单碱基突变,而遗传转化检测结果显示除了单碱基突变外,还有多碱基缺失突变。出现这种结果的原因可能是因为供以Cas9蛋白切割反应的时间较短,或者由于设计的检测酶切位点ScaⅠ与PAM距离较远(5 bp)导致更多的突变没有被检测到,除此之外,也可能是检测样本量少所致。
Ito等[22]编辑调节番茄果实成熟的MADs盒转录因子RIN基因,结果在T0再生植株中Cas9切割位点处检测到单碱基插入和超过3 bp的缺失突变,并且试验设计的3个靶点均发生编辑,而本研究中并未发现有碱基插入现象。综上所述,本研究对加工番茄α-Man的编辑效率低也可能与设计的sgRNA所含GC含量较低或个数较少有关。由于单个gRNA诱导的DSB可能被准确修复,导致编辑效率降低,因此,试验后期选择金门克隆(Golden gate cloning)这一快速、简便、全面的模块化载体构建系统[31],以pDIRECT-23C为载体骨架,构建双靶点敲除载体pDIRECT-Man。载体设计串联了2个gRNA诱导Cas9蛋白进行切割,来达到实现加工番茄α-Man的多重编辑,提高编辑效率的目的。除此之外,对于载体pDIRECT-Man的转基因植株,可以直接通过PCR的方式检测靶向缺失,使得α-Man突变体的筛选更加容易。目前,本研究部分处于加工番茄遗传转化阶段,后期转基因植株的获得和α-Man突变体的鉴定还有待进一步验证。
4 结论
成功在加工番茄中建立CRISPR/Cas9基因编辑体系,并实现对糖苷水解酶基因α-Man的编辑。
