ldentification of differentially expressed genes regulated by methylation in colon cancer based on bioinformatics analysis
2019-07-24YuLiangChengZhangDongQiuDai
Yu Liang, Cheng Zhang, Dong-Qiu Dai
Yu Liang, Cheng Zhang, Dong-Qiu Dai, Department of Gastrointestinal Surgery, the Fourth Affiliated Hospital of China Medical University, Shenyang 110032, Liaoning Province, China
Corresponding author: Dong-Qiu Dai, PhD, Chief Doctor, Professor, Surgical Oncologist,Department of Gastrointestinal Surgery, the Fourth Affiliated Hospital of China Medical University, No. 4 Chongshan East Road, Shenyang 110032, Liaoning Province, China.daidq63@163.com
Abstract BACKGROUND DNA methylation, acknowledged as a key modification in the field of epigenetics, regulates gene expression at the transcriptional level. Aberrant methylation in DNA regulatory regions could upregulate oncogenes and downregulate tumor suppressor genes without changing the sequences.However, studies of methylation in the control of gene expression are still inadequate. In the present research, we performed bioinformatics analysis to clarify the function of methylation and supply candidate methylation-related biomarkers and drivers for colon cancer.AIM To identify and analyze methylation-regulated differentially expressed genes(MeDEGs) in colon cancer by bioinformatics analysis.METHODS We downloaded RNA expression profiles, Illumina Human Methylation 450K BeadChip data, and clinical data of colon cancer from The Cancer Genome Atlas project. MeDEGs were identified by analyzing the gene expression and methylation levels using the edgeR and limma package in R software. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed in the DAVID database and KEGG Orthology-Based Annotation System 3.0, respectively. We then conducted Kaplan-Meier survival analysis to explore the relationship between methylation and expression and prognosis. Gene set enrichment analysis (GSEA) and investigation of protein-protein interactions (PPI) were performed to clarify the function of prognosis-related genes.RESULTS A total of 5 up-regulated and 81 down-regulated genes were identified as MeDEGs. GO and KEGG pathway analyses indicated that MeDEGs were enriched in multiple cancer-related terms. Furthermore, Kaplan-Meier survival analysis showed that the prognosis was negatively associated with the methylation status of glial cell-derived neurotrophic factor (GDNF) and reelin(RELN). In PPI networks, GDNF and RELN interact with neural cell adhesion molecule 1. Besides, GDNF can interact with GDNF family receptor alpha(GFRA1), GFRA2, GFRA3, and RET. RELN can interact with RAFAH1B1,disabled homolog 1, very low-density lipoprotein receptor, lipoprotein receptorrelated protein 8, and NMDA 2B. Based on GSEA, hypermethylation of GDNF and RELN were both significantly associated with pathways including “RNA degradation,” “ribosome,” “mismatch repair,” “cell cycle” and “base excision repair.”CONCLUSION Aberrant DNA methylation plays an important role in colon cancer progression.MeDEGs that are associated with the overall survival of patients may be potential targets in tumor diagnosis and treatment.
Key words: Colon cancer; Bioinformatics analysis; The Cancer Genome Atlas project;DNA methylation; Methylation-regulated differentially expressed genes; Overall survival
INTRODUCTION
Global cancer statistics for 185 countries reported that there were 1091601 new cases and 551269 deaths from colon cancer in 2018. The incidence and mortality were both ranked fourth worldwide[1]. Epidemiological studies have confirmed that the occurrence of colon cancer is related to multiple factors including smoking, alcohol abuse, insufficient activity, and high-fat food[2]. Although surgery-based comprehensive treatment is considered an effective treatment, a large number of patients still die from postoperative recurrence and metastasis[3]. Hence, research into specific biomarkers and therapeutic pathways is still of great value for improving patient prognosis.
Accumulating studies indicate that epigenetic modifications could promote the initiation and progression of colon cancer via multiple signaling pathways[4]. DNA methylation has been discovered and verified as a critical modification in the field of epigenetics. Numerous oncogenes and cancer-suppressor genes exhibit irregular expression, which is due to aberrant cytosine-phosphate-guanine (CpG)-island methylation in DNA regulatory regions rather than changes in the sequences[5]. For instance, the anti-oncogene ZNF350 undergoes epigenetic silencing due to hypermethylation of three sites in the promoter[6]. However, studies of DNA methylation in the individual genes remain inadequate. Identification of methylationregulated differentially expressed genes (MeDEGs) based on high-throughput data will be of profound significance for clarifying the role of methylation and identifying candidate directions for future research.
In recent years, with the development of gene-sequencing platforms, accumulating differentially expressed genes (DEGs) and epigenetic alterations have been revealed by bioinformatic analysis. For instance, Naumov et al[7]applied an Illumina Human Methylation 450K BeadChip to detect DNA methylation profiles in colorectal cancer tissues and colon tissues from cancer-free donors. As a result, 10342 hypermethylated and 5325 hypomethylated CpG sites were identified. Dumenil et al[8]further indicated that the Illumina Human Methylation 450K BeadChip could be utilized to detect and obtain high quality data from paraffin-embedded tissues. Multiple studies have shown that the Illumina Methylation 450K BeadChip has important application value in methylation research. Almost 450000 methylation sites and 96% of the CpG islands can be detected using the Illumina Human Methylation 450K BeadChip platform[9].However, there is still a lack of conjoint analysis of colon cancer methylation and studies on the correlation between methylation and patient prognosis in large cohorts.
In the present research, we downloaded in silico data and clinical data of colon cancer from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov)project[10]. MeDEGs were identified and related enrichment analysis was then performed. Moreover, we analyzed the correlation between the methylation status and overall survival of colon cancer patients. Protein-protein interaction (PPI)networks were constructed and gene set enrichment analysis (GSEA) of prognosisrelated MeDEGs was further performed.
MATERIALS AND METHODS
Sample collection
We downloaded colon cancer RNA expression profiles and clinical data from TCGA using the Genomic Data Commons Data Transfer Tool 1.3.0[10]. Up to December 2018,the public database included the expression profiles of 473 colon cancer tissues and 41 normal tissues (level 3) derived by RNA-seq. A total of 314 tumor tissues and 37 normal tissues had also been analyzed in the Illumina Human Methylation 450K BeadChip platform, and we also downloaded homologous methylation profiles from TCGA database. As 303 tumor tissues and 19 normal tissues were analyzed using both platforms, we merged the methylation and expression data together for correlation analysis. Based on the guidelines released by the National Cancer Institute in December 2015 (https://cancergenome.nih.gov/publications/publication guidelines), our research did not require the approval of an ethics committee.
Identification of MeDEGs
We constructed an RNA matrix and methylation data matrix containing the expression and methylation profiles using PERL software. Then, the coding gene IDs were converted to gene names based on information in the Ensembl database (Homo sapiens) (http://asia.ensembl.org/index.html). DEGs were identified with the edgeR package in R software with a threshold log2 fold change (FC) > 2.0 and P < 0.01.Moreover, differentially methylated genes (DMGs) were identified using the limma package with a threshold log2 FC > 1.0 and P < 0.01. Additionally, we analyzed the correlation between gene expression and methylation data through Spearman's correlation analysis. The upregulated-hypomethylated and downregulatedhypermethylated genes were identified as MeDEGs when they satisfied the cut-off criteria including correlation coefficient < -0.3 and P < 0.01. A volcano plot and heat map were drawn using R software.
In silico functional analysis of MeDEGs
To explore the function of the MeDEGs in colon cancer carcinogenesis and progression, gene ontology (GO)[11]enrichment analysis was performed using the DAVID database (https://david.ncifcrf.gov/). The GO enrichment analysis included three categories: cellular component, molecular function, and biological process.Furthermore, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways[12]were analyzed using KEGG Orthology-Based Annotation System 3.0 (http://koba s.cbi.pku.edu.cn/). Differences with P < 0.05 were regarded as statistically significant.
Association analysis between MeDEGs and patient prognosis
We divided the 314 colon cancer patients into two groups according to the median methylation value of the MeDEGs. Moreover, the patients were also divided into a hypermethylation and low-expression MeDEG (Hyper-LG) group and a hypomethylation and high-expression MeDEG (Hypo-HG) group according to the median value of methylation and expression of MeDEGs. Comparison of the overall survival between the two groups was then analyzed. Kaplan-Meier's method and the log-rank test were performed to assess survival rate. Differences with P < 0.05 were regarded as statistically significant.
GSEA and PPI network construction of prognosis-related MeDEGs
GSEA of prognosis-related MeDEGs was performed using GSEA 3.0 software with gene set c2 (cp.kegg.v.6.2.symbols.gmt). High throughput RNA expression of 303 colon cancer genes from TCGA were utilized as the dataset. Each sample was defined as either “H” or “L,” depending on whether it was greater than the median methylation value of prognosis-related MeDEGs or not. The number and type of permutations was set at “1000” and “phenotype,” respectively. An enrichment score >0.4 and P < 0.05 were regarded as statistically significant.
PPI analysis was conducted to reveal the molecular mechanisms of the prognosisrelated MeDEGs in colon cancer. We utilized the STRING protein database 11.0(http://string-db.org/) to construct the PPI networks. An interaction score > 0.4 was regarded as the cut-off criterion.
RESULTS
Identification of MeDEGs in colon cancer
First, we identified 1205 up-regulated and 892 down-regulated DEGs from 473 colon cancer and 41 normal tissues. The DEGs are shown as a volcano plot in Figure 1A.Second, 997 hypermethylated and 637 hypomethylated DMGs were identified from 314 colon cancer and 37 normal tissues. The heat map of the top 50 DMGs is shown in Figure 1B. Third, 514 genes were identified that showed a negative correlation between methylation and expression value. Finally, 5 up-regulated and 81 downregulated MeDEGs were identified that met the above three conditions. The Venn diagram and MeDEGs list are shown in Figure 2. The top 10 MeDEGs with the highest Spearman's correlation coefficients are shown in Figure 3.
Functional enrichment analyses of MeDEGs
To further investigate the function of MeDEGs in colon cancer, we subjected the 86 MeDEGs to GO and KEGG pathway analyses using DAVID 6.8. The enrichment analyses of GO are summarized in Table 1. A total of 13 biological processes, 5 cellular components, and 8 molecular functions were enriched among the MeDEGs. As for biological process and molecular function, “positive regulation of neuron differentiation” and “transcription factor binding” were the most enriched terms in the respective categories. KEGG pathway enrichment analysis suggested that MeDEGs predominantly participate in cancer-related pathways including“transcriptional misregulation in cancer”, “cAMP signal pathway”, and “cGMP-PKG signaling pathway” (Table 2).
MeDEGs related to the prognosis of colon cancer patients
We performed a Kaplan-Meier curve analysis to identify the MeDEGs related to overall survival of colon cancer patients. First, we analyzed the relationship between MeDEG methylation value and prognosis. The Kaplan-Meier curves showed that hypermethylation of glial cell-derived neurotrophic factor (GDNF) and reelin (RELN)were negatively correlated with overall survival (Figure 4A and B). Next, we further compared the prognosis of the above two MeDEGs between the Hyper-LG group and the Hypo-HG group. The results showed that Hyper-LGs were significantly related to poor survival of patients (Figure 4C and D). The expression, methylation data, and Spearman's correlation analyses of GDNF and RELN are shown in Figure 5.
GSEA and PPI network construction of GDNF and RELN
To clarify the biological function of GDNF and RELN methylation status, the effects of GDNF and RELN methylation status on KEGG pathways were analyzed using GSEA 3.0 software. The top ten GSEA results of GDNF and RELN are shown in Figure 6.Ultimately, hypermethylation of GDNF and RELN was both significantly associated with cancer-related pathways including “ribosome”, “RNA degradation”, “mismatch repair”, “cell cycle”, and “base excision repair”. More interestingly, the “ribosome”pathway was identified as the most significantly associated with GDNF and RELN methylation data.
Furthermore, we constructed PPI networks of GDNF and RELN using the STRING protein database. The PPI enrichment P-value was 1.74 × 10-5(Figure 7). The results revealed that GDNF and RELN interact with neural cell adhesion molecule 1.Moreover, GDNF can interact with several proteins including GDNF family receptor alpha (GFRA)-1, -2, and -3 and the proto-oncogene tyrosine-protein kinase receptor(RET). Additionally, RELN can interact with low-density lipoprotein receptor-related protein 8, glutamate receptor ionotropic, NMDA 2B, disabled homolog 1, very lowdensity lipoprotein receptor, and platelet-activating factor acetylhydrolase IB subunit alpha.
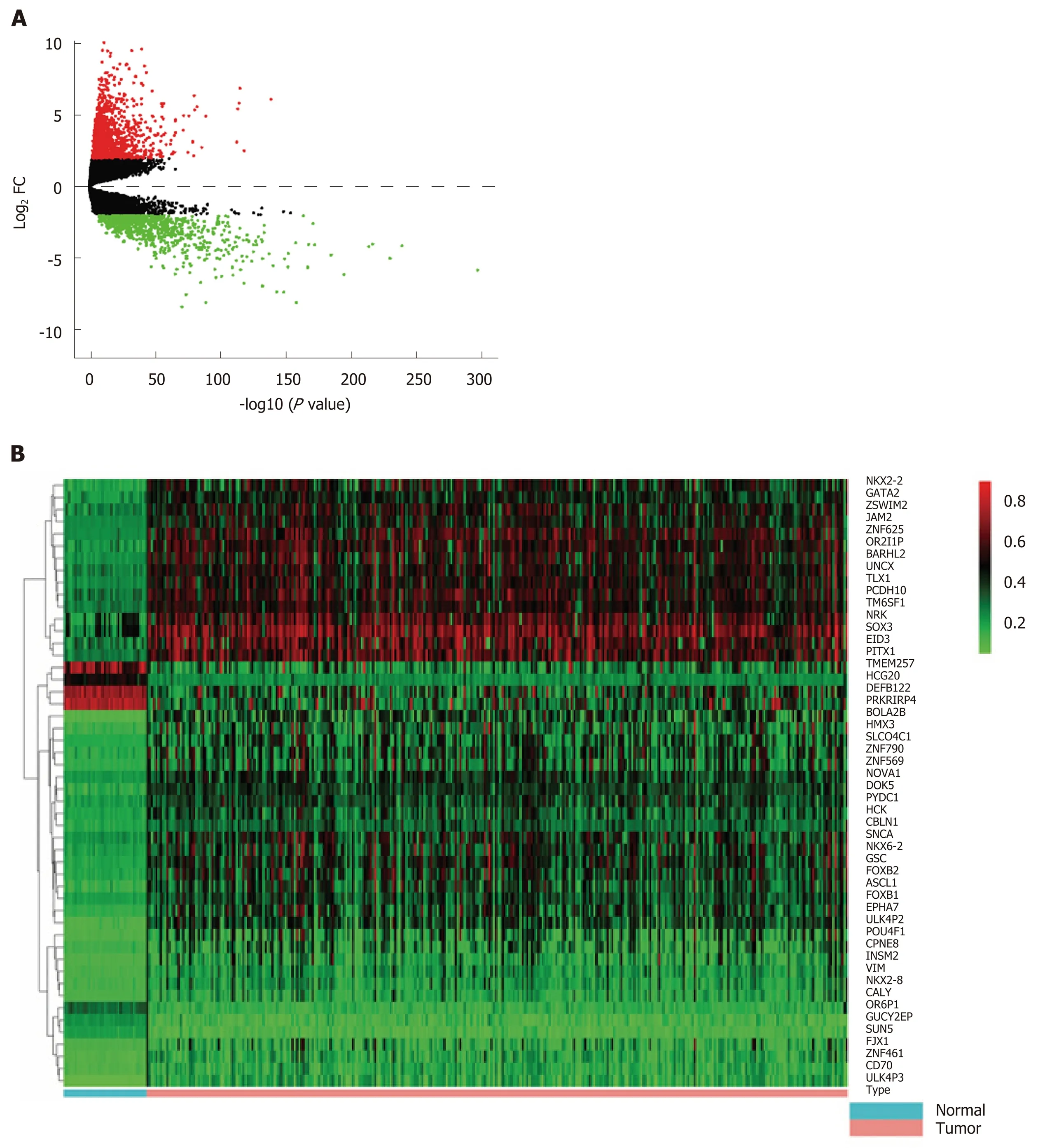
Figure 1 Differentially expressed genes and differentially methylated genes identified from The Cancer Genome Atlas project database. A: Volcano plot of differentially expressed genes between colon cancer and normal tissues [log2 fold change (FC) > 2, P < 0.01]. Red dots represent up-regulated genes and green dots represent down-regulated genes. Black dots represent the genes with a fold-change in expression of <2; B: Heat map of the top 50 differentially methylated genes(DMGs) (log2 FC > 1, P < 0.01). The left vertical axis shows clusters of DMGs and right vertical axis represents gene names. Red represents hypermethylated genes and green represents hypomethylated genes. DMGs: Differentially methylated genes; FC: Fold change.
DISCUSSION
Molecular pathological epidemiology (MPE), integrating molecular pathology and data science, can use tumor markers as surrogate of disease pathologies and help precision medicine. MPE deeply studies environmental exposures, intermediate variables, and molecular changes in cancer[13-15]. DNA methylation has been widely recognized as an important cancer-related biomarker and potential therapeutic target[16,17]in MPE. Previous studies mainly focused on the correlation between individual genes and methylation, so there was a lack of systematic analysis of DNA methylation in colon cancer. Thus, identification and analysis of MeDEGs in large cohorts of colon cancer patients are urgently required, providing potential directions and targets for future research.
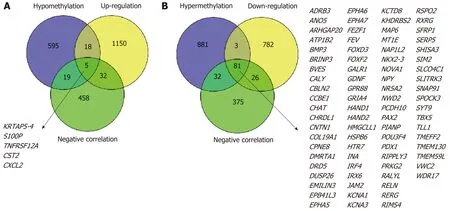
Figure 2 ldentification of methylation-regulated differentially expressed genes. A: A total of five genes were identified as methylation-regulated differentially expressed genes (MeDEGs) by taking the intersection of three gene sets (hypomethylation, up-regulation, and negative correlation); B: A total of 81 genes were identified as MeDEGs by taking the intersection of three gene sets (hypermethylation, down-regulation, and negative correlation). MeDEGs: Methylation-regulated differentially expressed genes.
In the present research, we identified a total of 5 up-regulated and 81 downregulated MeDEGs by integrating DEGs, DMGs, and the results of Spearman's correlation analysis. Among the 86 MeDEGs we report in this article, some of them have been confirmed to be regulated by methylation in previous research. For instance, the promoter of forkhead box D3 (FOXD3) has been reported to be hypermethylated in colon cancer and FOXD3 expression could be restored by treatment with 5-azacytidine[18,19]. The promoter methylation status of protocadherin 10 was related to disease-free survival and overall survival of colorectal patients,suggesting that it could be utilized as a biomarker for patients and to facilitate treatment decisions in colorectal cancer[20-22]. However, the methylation regulation mechanisms of the majority of MeDEGs have not been revealed and need to be further studied.
We further performed functional enrichment analysis to clarify the role of methylation in colon cancer. In biological process enrichment of GO, as many as 12 terms were related to the regulation of transcription. Therefore, we speculated that transcription factors regulated by methylation can further regulate the transcription of cancer-related genes and thus affect the occurrence and development of cancer.Aberrant cell proliferation is regarded as the distinguishing feature of cancer cells as opposed to normal cells. The term “negative regulation of cell proliferation” was enriched, meaning that multiple cancer suppressor genes were down-regulated by promoter hypermethylation. As shown in Table 1, MeDEGs including FEZ family zinc finger protein 1 (FEZF1), T-box transcription factor 5 (TBX5), secreted frizzled-related protein 5 (SFRP5), RAS-like estrogen regulated growth inhibitor (RERG), ripply transcriptional repressor 3 (RIPPLY3), and pancreatic and duodenal homeobox 1(PDX1) were involved. Among them, TBX5[23], SFRP5[24,25], RERG[26-28], and PDX1[29]have been reported to be regulated by methylation in colon cancer, while FEZF1 and RIPPLY3 have not been reported. Bone morphogenetic proteins (BMPs), which form part of the transforming growth factor β signaling pathway, can promote colon cancer cell migration and invasion[30,31]. Moreover, methylation of BMP3[32,33]and NK2 homeobox 5[34]promoters has been revealed as an independent biomarker for colon cancer. However, apart from colon cancer, there have been no reports on the role of CHRDL1 promoter region methylation in tumors[35]. KEGG pathway analysis has further clarified the function of MeDEGs. The “cAMP signaling pathway” has been indicated as one of the most critical mechanisms in regulating colon cancer cell apoptosis[36-38]. The term “transcriptional misregulation in cancer” is consistent with the GO enrichment results. Babykutty et al[39]indicated that “cGMP-PKG signaling pathway” could promote colon cancer cell migration and invasion by upregulating matrix metalloprotein 2/9.
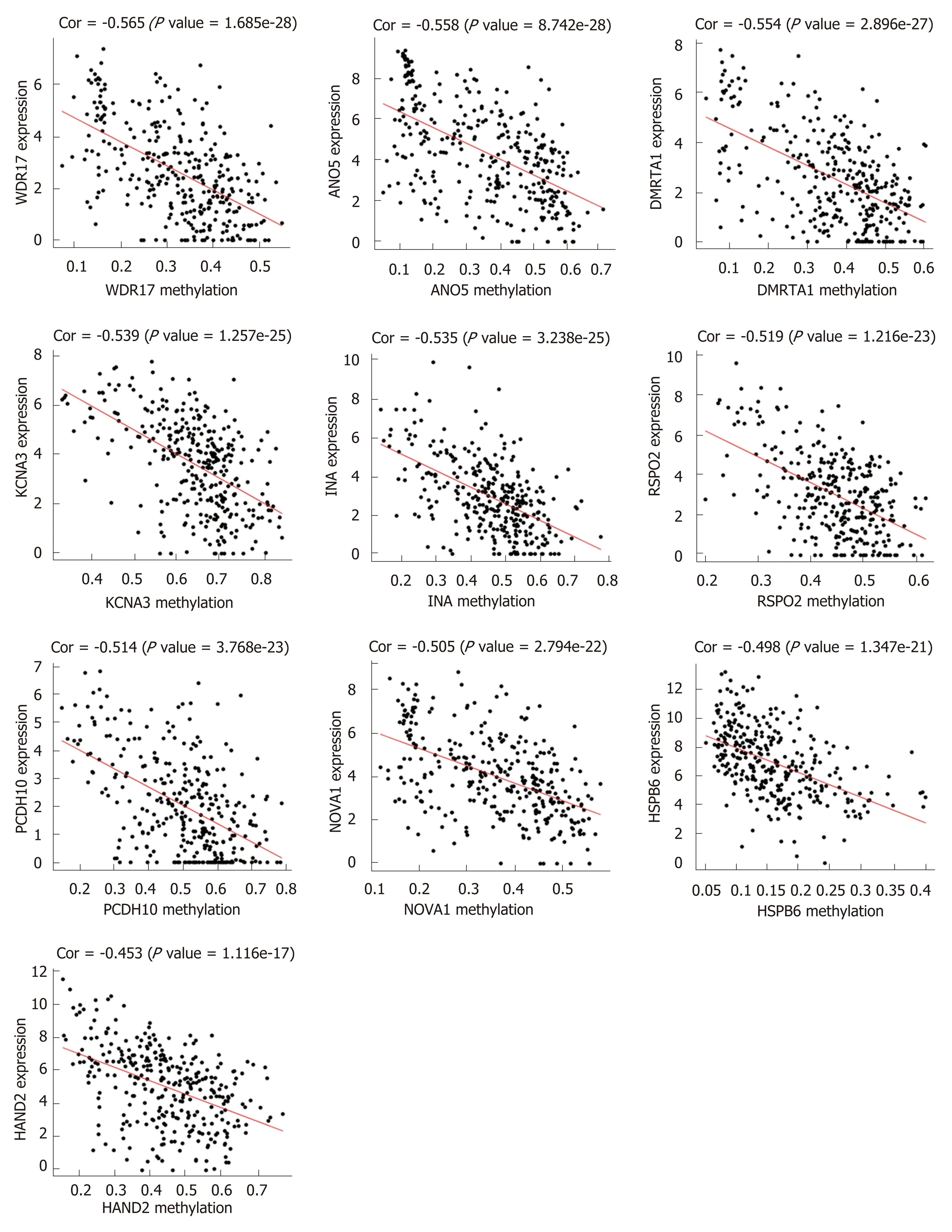
Figure 3 The methylation-regulated differentially expressed genes with the top 10 correlation coefficients. Spearman's correlation analysis was performed between methylation (horizontal axis) and expression (vertical axis) of methylation-regulated differentially expressed genes. Spearman's correlation coefficient and Pvalues are shown in each plot.
Moreover, we identified the methylation status of GDNF and RELN which was associated with clinical prognosis. GDNF is a neurotrophic factor which could guarantee the function of neuron in nervous systems[40]. According to TCGA database,GDNF is downregulated in colon cancer tissues. Interestingly, recent research indicated that GDNF promotes colon cancer cell proliferation and migration[41,42]. Luo et al[43]reported that GDNF was only detected in normal colon mucosa. Moreover,GDNF blocked apoptosis and suppression of anchorage-independent growth effects mediated by RET (rearranged during transfection). However, there has been no study on the correlation between GDNF methylation and the prognosis of colon cancer.RELN has been reported to play a critical role in maintaining epithelial cell ho-meostasis and protecting the colon from pathological processes[44]. Furthermore,RELN is epigenetically regulated by methylation in gastric cancer[45], pancreatic cancer[46], hepatocellular cancer[47], and myeloma[48]. However, the methylation state and function of RELN in colon cancer are still controversial.
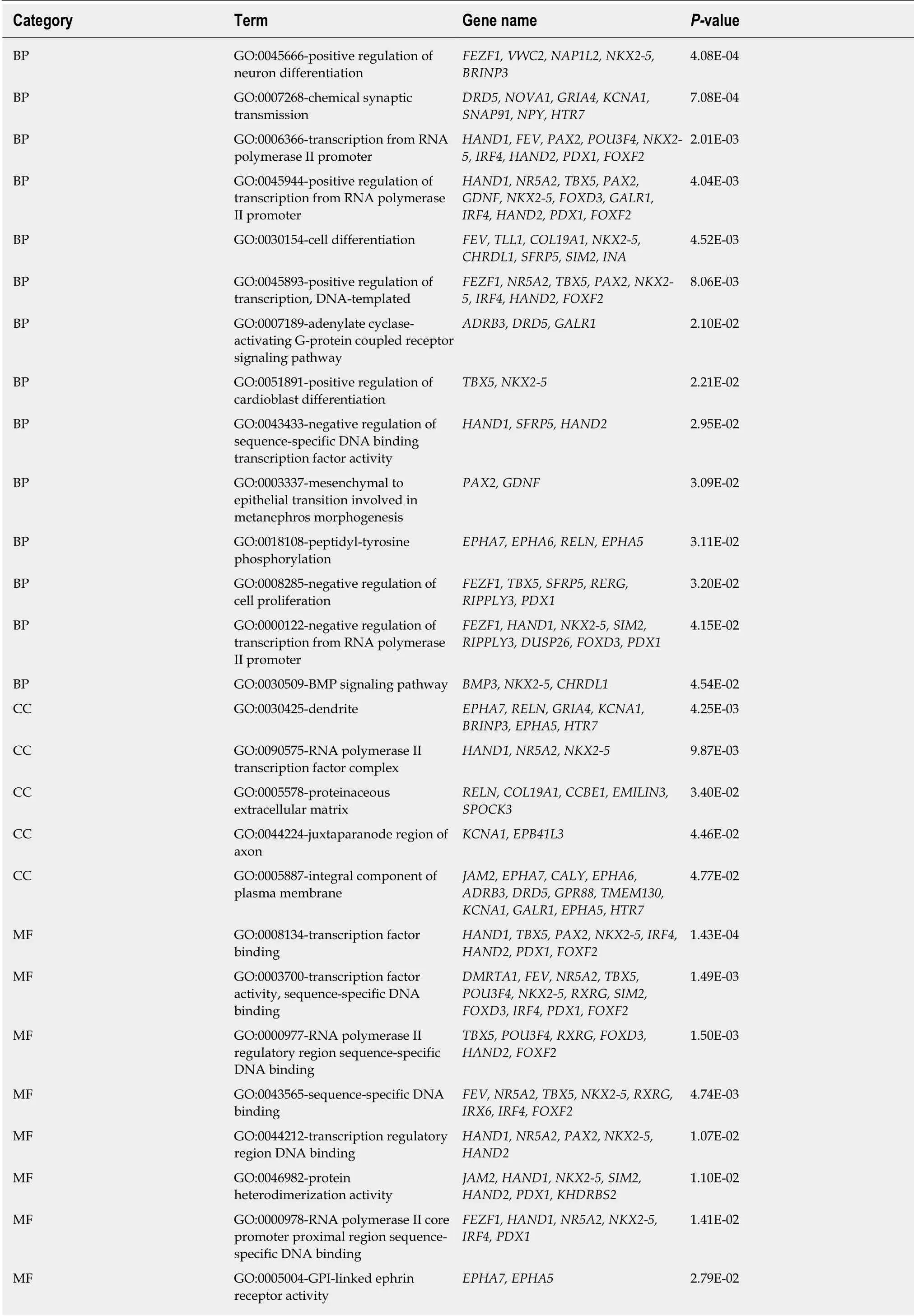
Table 1 Gene ontology enrichment analysis of methylation-regulated differentially expressed genes associated with colon cancer
Next, we performed GSEA to clarify the function of GDNF and RELN in KEGG pathways. The “ribosome” pathway is the most significantly enriched, which has been confirmed as an important mechanism in colon cancer development. For instance, precursor 45s ribosomal RNA (pre-45s rRNA) is up-regulated in colon cancer tissues and cell lines, and is associated with the prognosis of tumor patients.Pre-45s rRNA could promote cancer cell proliferation by inhibiting P53 by interfering in the interaction between murine double minute 2 (MDM2) and ribosomal protein L11 (RPL11)[49]. P73 is a p53 family tumor suppressor, which is promoted by RPL26 through direct binding to the 3'-UTR region. Furthermore, RPL26 maintains the protein stability of P73 by interacting with MDM2[50]. The “mismatch repair” and“base excision repair” pathways are primary mechanisms in DNA lesion repair and microsatellite instability in colon cancer[51]. Moreover, microsatellite-instability has been demonstrated to frequently occur in the hypermethylated cases. For instance,MLH1 (DNA mismatch repair protein Mlh1) methylation is a frequent molecular event and is closely linked to tumor invasion in veins and microsatelliteinstability[52,53]. Moreover, “base excision repair” capacity[54]and “cell cycle”[55]pathway are also the critical mechanism in colon cancer progression. From the above, the methylation state and function of GDNF and RELN should be better elucidated and replicated in a larger validation cohort.
In conclusion, we identified MeDEGs by analyzing the expression profiles and methylation data of colon cancer samples from TCGA database. Functional enrichment analyses further confirmed the role of MeDEGs in colon cancer. Moreover,we revealed that GDNF and RELN are related to overall survival. GSEA and PPI networks further clarified the function of prognosis-related MeDEGs. Our study deepens the understanding of methylation and provides novel therapeutic targets and prognosis-related biomarkers for further research.
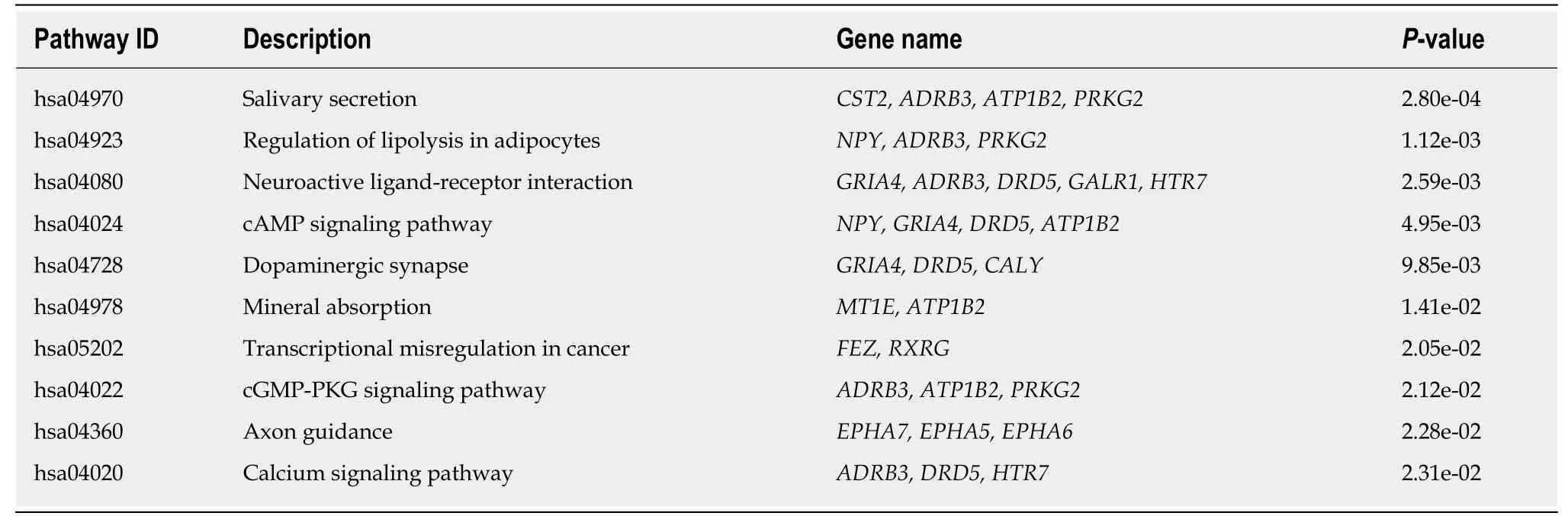
Table 2 Kyoto encyclopedia of genes and genomes pathway analysis of methylation-regulated differentially expressed genes associated with colon cancer
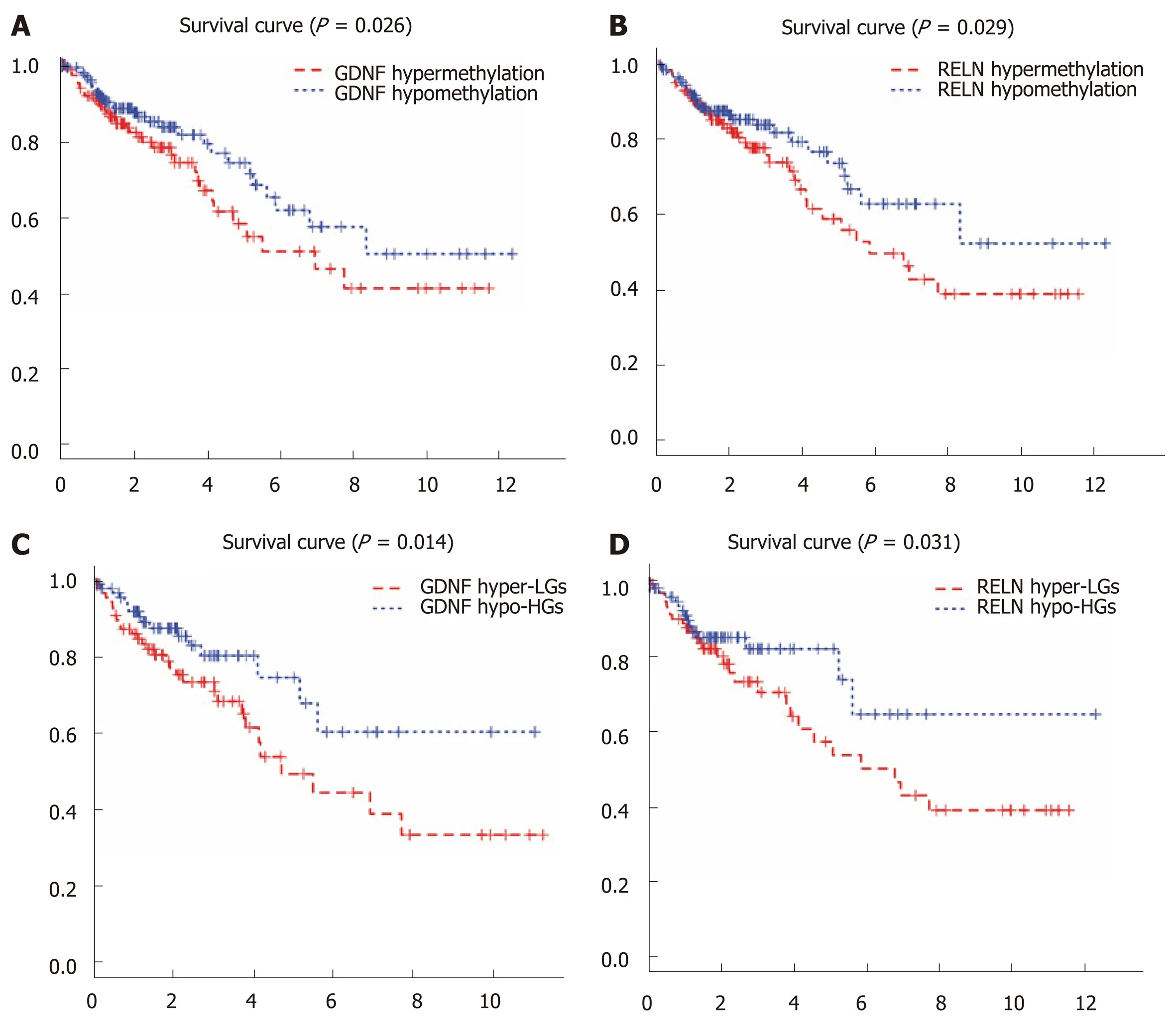
Figure 4 Kaplan-Meier curves for the methylation and methylation-expression of glial cell-derived neurotrophic factor and reelin. A and B: Glial cell-derived neurotrophic factor (GDNF) and reelin (RELN) were ranked by the median of methylation and then scored for each colon cancer patient in accordance with high- or low-level methylation value; C and D: GDNF and RELN were ranked by the median of methylation and expression and then scored for each colon cancer patient in accordance with high- or low-level methylation value and high or low-level expression value. The horizontal axis represents the overall survival time and the vertical axis represents survival function. Hyper-LGs: Hypermethylation and low expression methylation-regulated differentially expressed genes; Hypo-HGs: Hypomethylation and high expression methylation-regulated differentially expressed genes; GDNF: Glial cell-derived neurotrophic factor; RELN: Reelin.
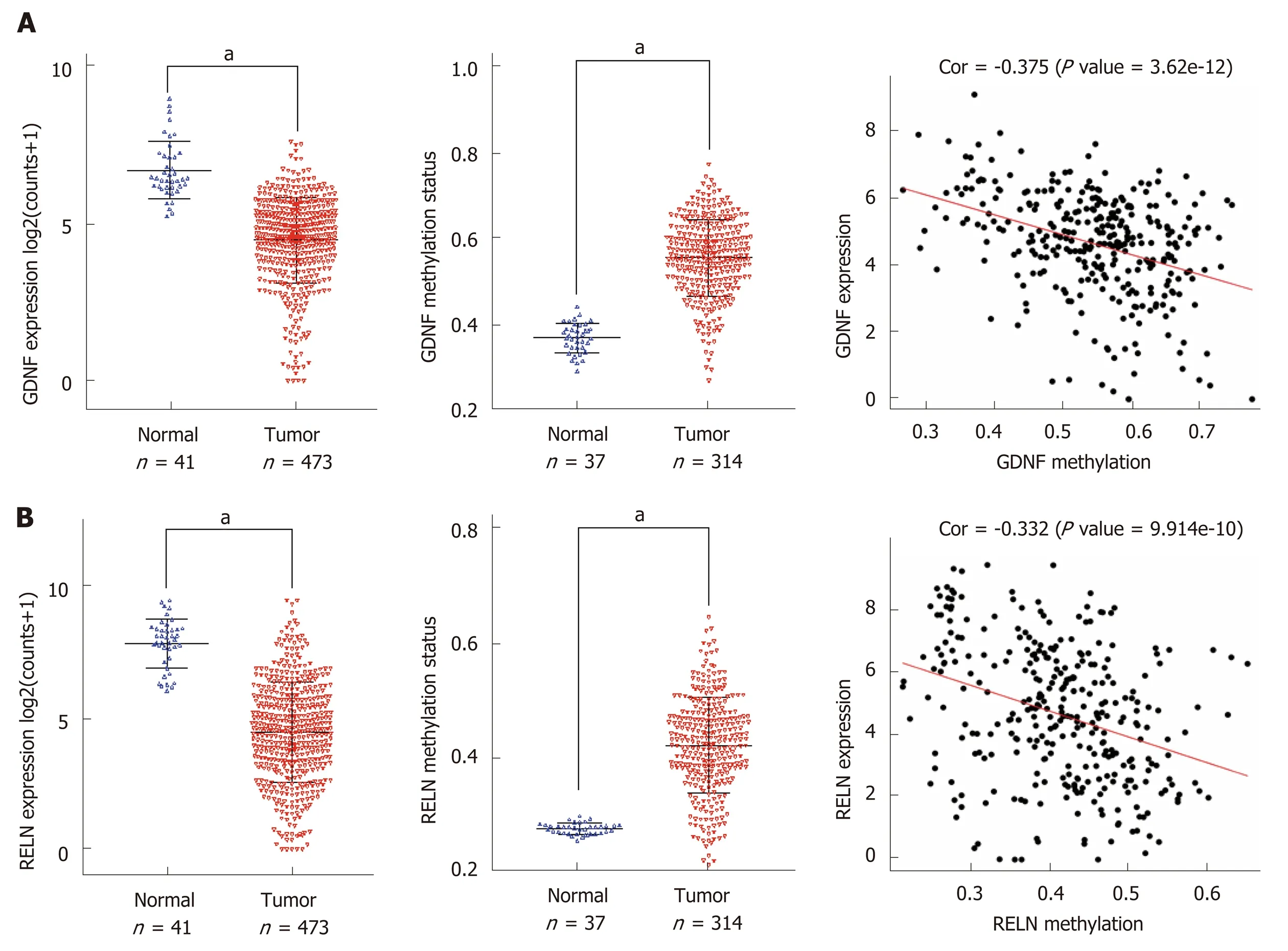
Figure 5 The expression, methylation status, and Spearman's correlation analysis of glial cell-derived neurotrophic factor and reelin. A: The Cancer Genome Atlas (TCGA) database was utilized to analyze the expression and methylation status of glial cell-derived neurotrophic factor and the correlation between them; B: The TCGA database was utilized to analyze the expression and methylation status of reelin and the correlation between them. aP < 0.01. GDNF: Glial cellderived neurotrophic factor; RELN: Reelin; TCGA: The Cancer Genome Atlas.
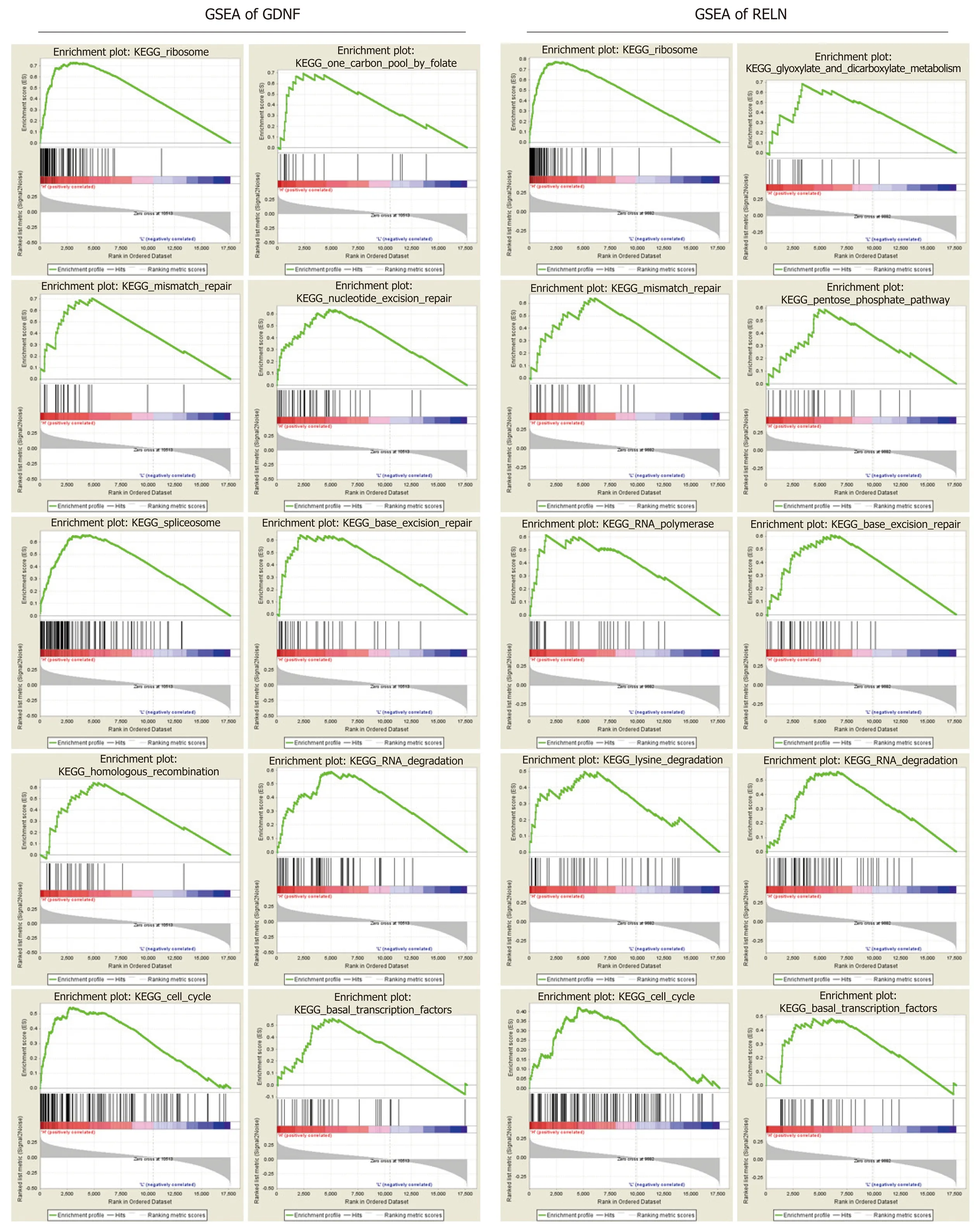
Figure 6 Gene set enrichment analysis of glial cell-derived neurotrophic factor and reelin. A: Gene set enrichment analysis (GSEA) of glial cell-derived neurotrophic factor (GDNF); B: GSEA of reelin (RELN). GSEA of GDNF and RELN showing that hypermethylation of GDNF and RELN were enriched in multiple cancer-related pathways. GDNF: Glial cell-derived neurotrophic factor; RELN: Reelin; GSEA: Gene set enrichment analysis; KEGG: Kyoto Encyclopedia of Genes and Genomes.
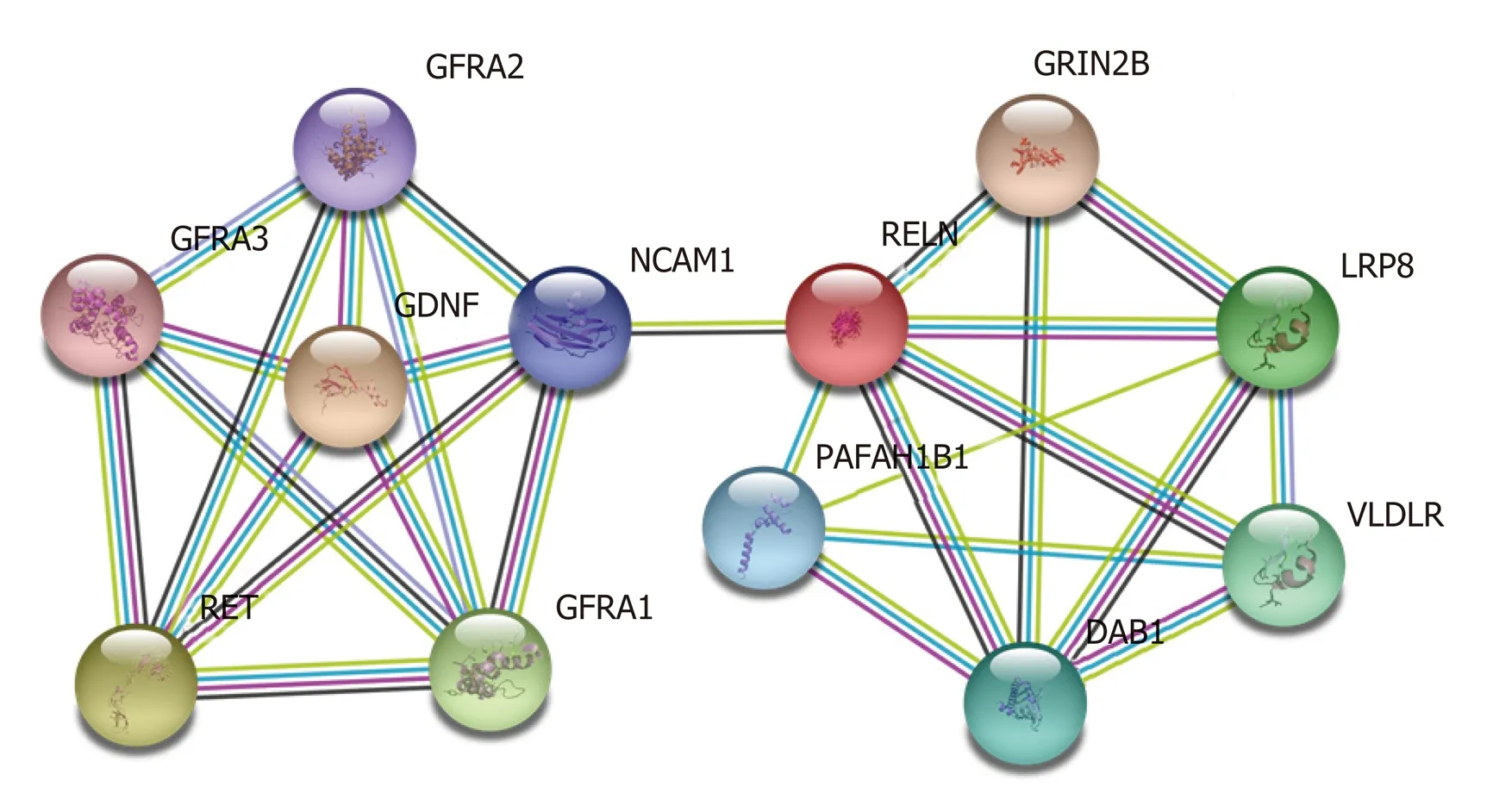
Figure 7 Protein-protein interactions of glial cell-derived neurotrophic factor and reelin. The STRING protein database was utilized to analyze the proteinprotein interactions of glial cell-derived neurotrophic factor and reelin.
ARTICLE HIGHLIGHTS
Research background
Accumulating evidence has indicated that DNA methylation modification is a reversible process of gene regulation in epigenetics. However, studies of methylation in the individual genes and pathways are still insufficient. In the present research, we conducted a conjoint analysis of correlation between methylation and gene expression and patient prognosis in large cohorts based on the Illumina Methylation 450K BeadChip.
Research motivation
DNA methylation modification has been considered as a potential therapeutic target and biomarker that may improve the prognosis of colon cancer. Therefore, identification and analysis of methylation-regulated differentially expressed genes (MeDEGs) will be of great significant.
Research objectives
In our study, we aimed to conduct bioinformatics analysis to identify MeDEGs and prognosisrelated MeDEGs in colon cancer. Functional enrichment analysis was performed to clarify the function of MeDEGs. Furthermore, our study elucidated the potential mechanisms of prognosisrelated MeDEGs.
Research methods
We downloaded RNA expression profiles, Illumina Human Methylation 450K BeadChip data,and clinical data of colon cancer from The Cancer Genome Atlas project. Differentially expressed genes and differentially methylated genes were identified using with the “edgeR” package and the “limma” package in R software. Then, we performed Spearman's correlation analysis to clarify the relationship between methylation and expression. The in silico function of MeDEGs was further analyzed in the DAVID database and Kyoto Encyclopedia of Genes and Genomes(KEGG) Orthology-Based Annotation System 3.0, respectively. The relationship between methylation and expression and overall survival was revealed through a Kaplan-Meier curve test. Gene set enrichment analysis (GSEA) and investigation of protein-protein interactions were performed to clarify the function of prognosis-related genes.
Research results
We identified a total of 5 up-regulated and 81 down-regulated MeDEGs that satisfied the conditions. Gene ontology analysis indicated that the enrichment terms are mainly associated with transcription regulation. According to KEGG pathway analysis, three cancer-related pathways were involved by MeDEGs. Hypermethylation of glial cell-derived neurotrophic factor(GDNF) and reelin (RELN) was negatively correlated with overall survival. Based on GSEA,hypermethylation of GDNF and RELN was both significantly associated with pathways including “RNA degradation,” “ribosome,” “mismatch repair,” “cell cycle”, and “base excision repair.”
Research conclusions
In conclusion, we provide a new and reliable pathway to identify MeDEGs based on Illumina Human Methylation 450K BeadChip. Methylation plays a critical role in regulating cancerrelated gene expression, especially tumor-suppressor genes. Our study provides an in-depth understanding of methylation. Furthermore, the prognosis-related MeDEGs may be potential biomarkers and therapeutic targets in colon cancer.
Research perspectives
The application of high-throughput platform will provide great support for the precision medicine in cancer. We will conduct more studies to reveal the function of the prognosis-related MeDEGs and establish a valid and reliable high-throughput analysis system in the future.
杂志排行

World Journal of Gastroenterology的其它文章
- Diuretic window hypothesis in cirrhosis: Changing the point of view
- Fluoroquinolones for the treatment of latent Mycobacterium tuberculosis infection in liver transplantation
- Reactivation of hepatitis B virus infection in patients with hemolymphoproliferative diseases, and its prevention
- Current status of endoscopic retrograde cholangiopancreatography in patients with surgically altered anatomy
- Choledochal cysts: Similarities and differences between Asian and Western countries
- Gastro-duodenal disease in Africa: Literature review and clinical data from Accra, Ghana