基于分子对接方法设计新的喹啉类表皮生长因子受体抑制剂
2019-07-18彭鹏辉薛艾奇
刘 丹, 张 毅, 彭鹏辉, 薛艾奇
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
表皮生长因子受体 (epidermal growth factor receptor,EGFR) 家族属于受体酪氨酸激酶家族,包括 EGFR (HER1/ErbB-1)、ErbB-2 (HER2/neu)、ErbB-3 (HER-3)和 ErbB-4 (HER-4).EGFR型酪氨酸激酶抑制剂可以进入细胞内,直接作用于EGFR 家族受体的胞内区,干扰ATP结合,阻断激酶的自身磷酸化,从而阻断异常的信号传导产生抑瘤效果.目前 FDA 批准上市的药物有吉非替尼 (gefitinib,Iressa,2003)、厄洛替尼 (erlotinib,Tarceva,2004)、拉帕替尼(lapatinib,Tykerb,2007) 和阿法替尼 (afatinib,Gilotrif,2013)[1].这4种激酶抑制剂均具有喹唑啉骨架结构.
与喹唑啉结构相似的喹啉类化合物也是一类重要的杂环衍生物,以其独有的化学结构对许多激酶有抑制作用,具有显著生理活性[2].在喹唑啉类化合物的构效关系基础上设计喹啉结构酪氨酸激酶抑制剂,如美国Wyeth制药公司开发的3-氰基喹啉类不可逆酪氨酸激酶抑制剂来那替尼(Neratinib),已经进入三期临床[3-4].本文将8个7-甲基-4-苯氨基喹啉化合物与EGFR进行对接,以Neratinib为阳性对照,根据抑制剂和受体的作用模式及结合能结果设计一个新的喹啉化合物分子,为进一步设计合成新型的 EGFR 酪氨酸激酶抑制剂提供参考.
1 实验部分
参考朱丽荔等的《表皮因子受体和抑制剂之间的分子对接的研究》[5]、康从民等的《五元杂环并嘧啶类胸苷酸合成酶抑制剂的构效关系和分子对接》[6]、朱瑞新主编的《计算机辅助药物设计:基本方法原理概要与实践详解》[7]以及所学知识设计实验.
1.1 蛋白质结构的准备
2gs7是从蛋白数据库里获得EGFR与AMP-PNP的复合物[8].在分子对接前先除去晶体中原有的水分子,并对其原来的最佳配体做出球心位置的记录,将2gs7复合物中原有配体除去,得到用于分子对接的EGFR酪氨酸激酶结构.
1.2 分子对接
处于Ⅲ期临床的抑制剂来那替尼具有喹啉母核,故选取来那替尼(图1)为阳性对照.用Molegro Virtual Docker软件分别对来那替尼和8个7-甲基-4-苯氨基喹啉化合物X1~X8(图2)与EGFR进行独立对接,选取结合能最低的构象为最佳构象,最后运用Discovery Studio 2016 Client软件判断化合物和受体作用模式.

图1 来那替尼结构

图2 7-甲基-4-苯氨基喹啉化合物结构
2 结果与讨论
2.1 分子对接的结合能
用Molegro Virtual Docker对来那替尼和化合物X1~X8进行分子对接,得到结合能(Eb),如表 1所示.
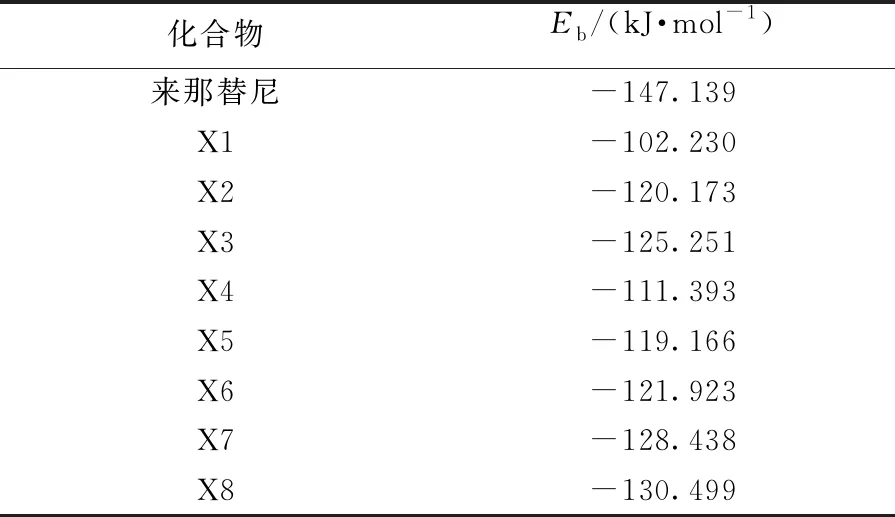
表1 来那替尼、X1~X8与EGFR对接的结合能(Eb)
2.2 结合位点分析
小分子抑制剂与酶靶向结合时,主要是和抑制剂周围一定距离内的氨基酸残基相互作用,这些残基可以被认为是活性残基[6].选择结合能较小的X7、X8和来那替尼,运用Discovery Studio 2016 Client软件分析其与受体的相互作用.
图3显示,化合物X7的4位苯氨基N上的H伸入活性口袋中,与氨基酸残基ASP831形成氢键;苯环上的4′位NO2中的O与LEU753、PHE832形成氢键作用.喹啉环与THR766、LEU764形成碳氢键.

图3 化合物X7与EGFR相互作用的三维模型
图4显示,化合物X8的4位苯氨基N上的H伸入活性口袋中,与氨基酸残基ASP831形成氢键;4′位OCH3上的O与LEU753形成氢键;喹啉环2位上的C与氨基酸残基LEU764形成碳氢键;喹啉环上的π键与THR766形成碳氢键.
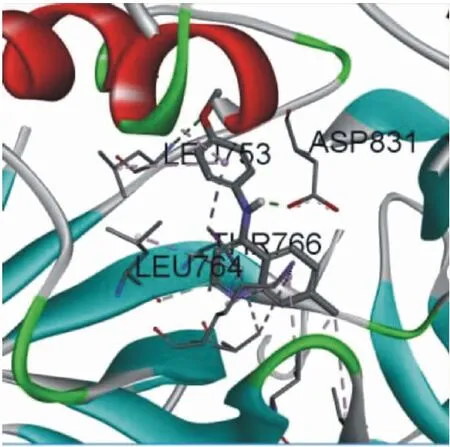
图4 化合物X8与EGFR相互作用的三维模型
来那替尼 4 位芳氨基团位于受体的疏水性口袋中[9],从图5可以看出:来那替尼喹啉环上4位苯氨基N上的H与MET769形成氢键;6位的NH中的H与THR830形成氢键;7位上的O与THR766形成氢键.

图5 来那替尼与EGFR相互作用的三维模型
2.3 设计新化合物分子X9
由表1结合能数据、图1、图2化合物结构可以看出:X1的结合能最大,X1中R基都是H,4-苯氨基体积最小;X3中3′,4′位的H都被Cl取代,与X2相比,4位苯氨基所占体积更大,所得结合能更小;X4和X5之间也表现出相似的规律.化合物X7、X8与受体对接结合能更低(-128.438 kJ·mol-1、-130.499 kJ/mol-1),苯环4′位分别连接NO2、OCH3基团,NO2、OCH3官能团相对于卤素和氨基体积较大;来那替尼与受体对接结合能为-147.139 kJ/mol-1,苯环4′位连接的基团为吡啶-2-乙氧基.
X7、X8苯氨基4′位的NO2、OCH3官能团上的O均能与氨基酸残基LEU753形成氢键,据此,在化合物X6的4′氨基上引入烟酰基,增大4位苯氨基的体积,同时引入O原子,得到化合物X9(美国化学文摘中没有这个化合物),结构如图6所示.将化合物X9与EGFR对接,结合能为-136.933 kJ/mol.X9与EGFR形成的复合物如图7所示,喹啉环上4位N上的H与THR830形成氢键.结果显示,新设计的化合物喹啉环4位苯氨基体积增大,与受体结合能力得到增强,形成氢键多少并不是配体与受体亲和力强弱的决定性因素.
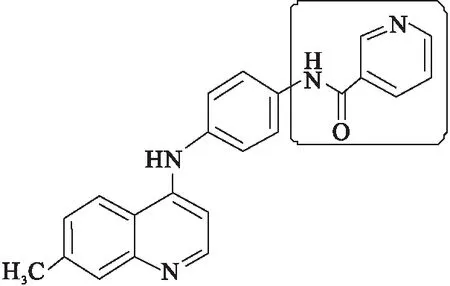
图6 化合物X9结构

图7 化合物X9与EGFR相互作用的三维模型
3 结 论
将来那替尼及7-甲基-4苯氨基喹啉化合物(X1~X8)与EGFR对接,计算结合自由能,分析与受体的作用模式.发现4′位官能团的体积增大,与受体结合能力增强.设计新化合物X9,X9与EGFR对接结合能为-136.933 kJ/mol,结合能力强于化合物X1~X8,喹啉环上4位N上的H与THQ830形成氢键.获得的喹啉衍生物结构新颖,可能成为抑制 EGFR酪氨酸激酶的先导化合物或候选药物,其生物活性有待进一步实验验证,本文结果为喹啉类EGFR酪氨酸激酶抑制剂的开发提供了参考.
