金属酞菁配合物催化的咪唑N-芳基化反应
2019-07-18李艳丽
蔡 捷, 李艳丽, 贺 旭, 顾 昊, 丁 茯
(沈阳化工大学 化学工程学院, 辽宁 沈阳 110142)
作为一类非常重要的有机合成中间体,N-芳基咪唑类在生物学、药学及N-杂环卡宾化学中有重要意义[1].常用合成方法有两种:亲核芳香取代反应和金属离子钯与铜参与的C-N偶联反应.前者需要芳卤上要有很强的吸电子基团,且反应需要极性溶剂、高温才能进行;而钯催化剂高昂的价格、较强的毒性以及对含磷配体的依赖造成其在交叉偶联反应中也不能广泛应用;铜催化剂虽然避免了上述缺陷,但是反应时间长,通常需要24 h以上[2-3].因此,寻找新的合成方法是有机合成化学家关注的热点之一.
金属酞菁配合物(M-Pc)具有廉价易得、热稳定性、化学稳定性良好等优点,常作为有机反应的重要催化剂[4].目前,金属酞菁化合物主要用于催化氧化反应,如Cu-Pc和Co-Pc催化氧化苯酚类物质[5-7]、Fe-Pc催化不饱和酮和水合肼的氧化-芳香化反应[8]等.另外,金属酞菁化合物还用于其他类型的催化反应,如Cu-Pc催化烯烃的环丙烷化反应[9]和催化醛、酮、腈一锅合成β-丙烯酰胺酮化合物[10],Fe-Pc和Mg-Pc催化CO2和环丙烷的环加成反应等[11].催化C-N偶联反应的应用目前只有一例.在该反应中,无取代的Cu-Pc被用于催化咪唑的N-芳基化反应,反应时间长,即使使用活性较高的芳基碘为底物,也需要24 h[12].该反应催化效率低的原因可能为酞菁的大平面分子结构分子间具有强烈的相互作用,导致其易于在反应液中形成二聚物或多聚物,从而会减少轴向配位的活性位点,进而导致催化效率降低、反应时间较长.为验证这种推测,破坏分子之间的聚集,从而大幅提高催化活性,本文合成了在α位上具有较大空间位阻芳氧基的铜酞菁配合物Cu-Pc-1和Cu-Pc-2,β位上具有芳氧基的铜酞菁配合物Cu-Pc-3和铁酞菁配合物Fe-Pc-3,并研究了这些金属酞菁配合物对咪唑N-芳基化反应的催化性能.
1 实验部分
1.1 主要试剂和仪器
3-硝基邻苯二腈(AR)、4-硝基邻苯二腈(AR)、尿素(AR)、1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU,AR)、咪唑(AR)、溴苯(AR)、KOH(AR)、无水硫酸钠(AR)、CuCl(AR)和FeCl3(AR),阿达玛斯试剂有限公司;8-羟基喹啉(AR)、4-叔丁基苯酚(AR)和钼酸铵(AR),上海麦克林生化科技有限公司;K2CO3(AR)、硝基苯(AR)、乙酸乙酯(AR)、二甲基亚砜(AR)和1-戊醇(AR),国药集团化学试剂有限公司.实验中溶剂均为分析纯.
Bruker ARX-500核磁共振仪(CDCl3溶剂,TMS内标);LCQ Deca XP液相色谱-质谱联用仪,美国热电-菲尼根公司;LC-20A液相色谱仪,岛津(中国)有限公司;NEXUS470红外光谱仪,美国热电-菲尼根公司;UV-Vis紫外可见光谱仪,岛津(中国)有限公司.
1.2 合成路线
合成路线如下所示:


1.2.1 前体邻苯二腈衍生物的合成
将3-硝基邻苯二腈(1.73 g,10 mmol)或4-硝基邻苯二腈(1.73 g,10 mmol)、4-叔丁基苯酚(1.50 g,10 mmol)或8-羟基喹啉(1.45 g,10 mmol)和K2CO3(2.76 g,20 mmol)加入到二甲基亚砜(DMSO,20 mL)中,并在氩气保护下室温反应10 h.反应结束后,将反应混合物倒入水(1000 mL)中并搅拌1 h,过滤,滤饼用大量的水洗,干燥得到粗品.硅胶柱层析(洗脱剂:CH2Cl2)得到纯的目标化合物.
3-(4-叔丁基苯氧基)邻苯二腈:白色的固体产物(2.29 g),产率83.0 %.1H NMR(500 MHz,CDCl3),δ:7.59(t,1H),7.46(m,3H),7.11(d,1H),7.04(d,2H),1.35(s,9H).
4-(4-叔丁基苯氧基)邻苯二腈:白色固体产物(2.10 g),产率76.1 %.1H NMR(500 MHz,CDCl3),δ:7.73(d,1H),7.48(d,2H),7.27(s,2H),7.01(d,2H),1.37(s,9H).
3-(8-喹啉氧基)邻苯二腈:白色固体产物(2.15 g),产率79.1 %.1H NMR(500 MHz,CDCl3),δ:8.85(d,1H),8.26(d,1H),7.82(d,1H),7.62(t,1H),7.53(d,1H),7.49(m,1H),7.45(d,2H),6.85(t,1H).
1.2.2 铜酞菁配合物的合成
1.2.1.1 钼酸铵催化法
将3-(8-喹啉氧基)邻苯二腈(3 390.9 mg,12.5 mmol)或3-(4-叔丁基苯氧基)邻苯二腈(3 454.1 mg,12.5 mmol)、CuCl(297.0 mg,3.0 mmol)、尿素(1 486.5 mg,24.8 mmol)和钼酸铵(58.8 mg,0.3 mmol)加入到硝基苯(12 mL)中,并在氩气保护下160 ℃搅拌10 h.反应结束后,减压蒸馏除去溶剂.残余物用硅胶柱层析得到目标产物.
Cu-Pc-1:硅胶柱层析[洗脱剂:v(CHCl3)∶v(MeOH)=100∶1]得到蓝绿色粉末(1.29 g),摩尔产率37.3 %.ESI-MS(m/z):1 149.0[M+H]+(calcd for C68H37CuN12O4,1 148.2).元素分析(C68H36CuN12O4,计算值/实测值),w/%:C,71.10/70.83;H,3.16/3.29;N,14.63/14.34.
Cu-Pc-2:硅胶柱层析(洗脱剂:CHCl3)得到蓝绿色粉末(1.44 g),产率41 %.ESI-MS(m/z):1 168.3[M+H]+(calcd for C72H65CuN8O4,1 168.5).元素分析(C72H64CuN8O4,计算值/实测值),w/%:C,73.98/74.22; H,5.52/5.79; N,9.59/9.41.
1.2.2.2 DBU催化法
将4-(4-叔丁基苯氧基)邻苯二腈(3 454.1 mg,12.5 mmol)、CuCl(297.0 mg,3.0 mmol)或FeCl3(486.6 mg,3.0 mmol)和DBU(1 mL)加入到正戊醇(12 mL)中,并在氩气保护下135 ℃搅拌16 h.反应结束后,减压蒸馏除去溶剂.残余物用热的甲醇和水反复洗涤,剩余物用硅胶柱层析(洗脱剂:CHCl3)得到纯的目标化合物.
Cu-Pc-3:蓝绿色粉末(1.12 g),产率32 %.ESI-MS(m/z):1 169.0[M+H]+(calcd for C72H65CuN8O4,1 168.5).元素分析(C72H64CuN8O4,计算值/实测值),w/%:C,73.98/74.16; H,5.52/5.73; N,9.59/9.88.
Fe-Pc-3:蓝色粉末(1.12 g),产率 32 %.
ESI-MS(m/z):1 161.7[M+H]+(calcd for C72H65FeN8O4,1 161.4).元素分析(C72H64FeN8O4,计算值/实测值),w/%:C,74.47/74.17; H,5.56/5.42; N,9.65/9.89.
1.3 铜酞菁催化咪唑的N-芳基化反应
金属酞菁配合物催化的咪唑N-芳基化反应如下所示:

催化步骤:将咪唑(306.4 mg,3.0 mmol)、溴苯(471.0 mg,3.0 mmol)、KOH(336.6 mg,6 mmol)和金属酞菁催化剂(摩尔分数0.67 %)加入到DMSO(4 mL)中,并在氩气保护下110 ℃反应.薄层色谱层析和HPLC跟踪反应.反应结束后,反应液倒入蒸馏水中,用乙酸乙酯萃取3次.合并的有机相用无水Na2SO4干燥,过滤并浓缩.残余物用硅胶柱层析(洗脱剂:乙酸乙酯)得到目标产物N-苯基咪唑.1H NMR(500 MHz,CDCl3),δ:7.77(s,1H),7.36(d,2H),7.27(d,3H),7.19(s,1H),7.11(s,1H).
2 结果与讨论
2.1 红外光谱分析
由表1和图1可以看出:4种金属酞菁配合物的IR谱图很相似.
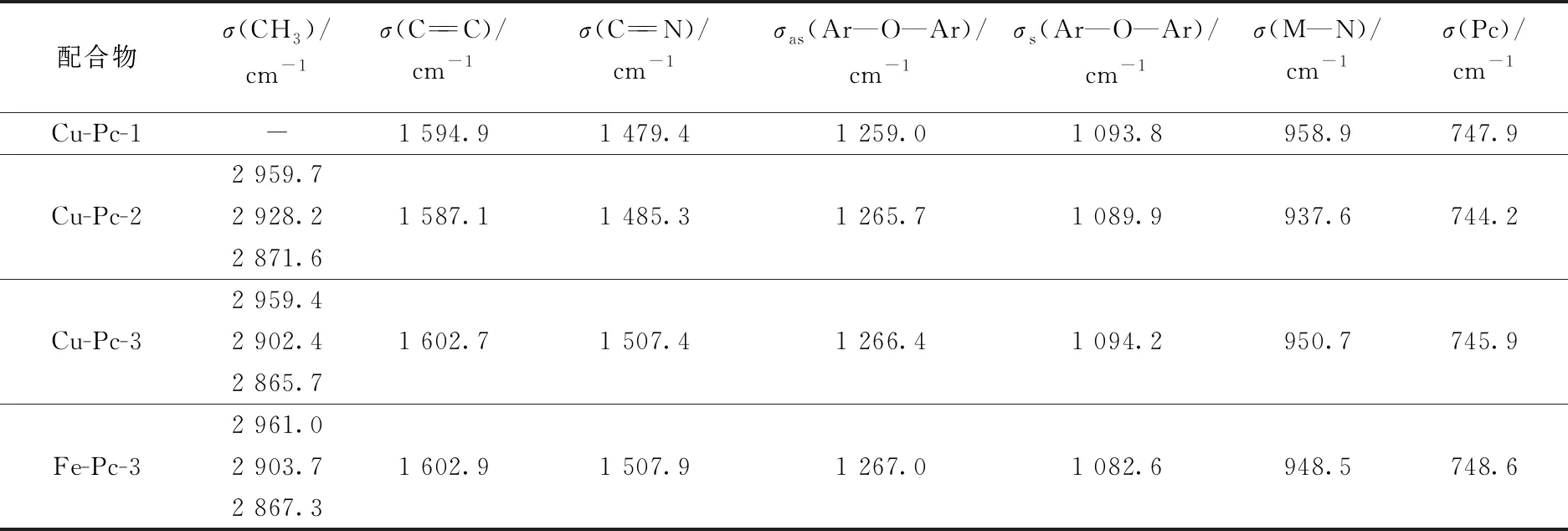
表1 金属酞菁配合物的部分特征IR光谱数据及其归属

图1 金属酞菁配合物的IR谱图
以Cu-Pc-2为例,2 959.7、2 928.2和2 871.6 cm-1处的吸收峰为甲基的对称和不对称伸缩振动;1 587.1和1 485.3 cm-1处的吸收峰分别为酞菁环的C==C和C==N伸缩振动,即酞菁环的骨架振动;1 265.7和1 089.9 cm-1处的吸收峰分别为芳醚键的不对称和对称伸缩振动;937.6 cm-1处的吸收峰可归属为金属-配体振动吸收峰;744.2 cm-1处的吸收峰可归属为酞菁环的振动吸收.而对于不含甲基的Cu-Pc-1,在2 900 cm-1附近处没有甲基的吸收峰.
2.2 紫外可见光谱分析
以α位取代的Cu-Pc-2和β位取代的Cu-Pc-3为例,研究金属酞菁配合物上取代基引入的位置不同对紫外可见光谱的影响.如图2所示,在CHCl3中,2个铜酞菁配合物在紫外可见光区有2个特征吸收带:B带在318 nm附近的近紫外区,Q带在714 nm附近的可见光区.比较这两个铜酞菁配合物的紫外可见光谱,可以发现取代基的位置不同对金属酞菁配合物在B带和Q带的最大吸收波长几乎没有影响.

图2 金属酞菁配合物在CHCl3中的UV-Vis光谱
2.3 金属酞菁配合物催化的咪唑N-芳基化反应
以咪唑和溴苯为模板反应考察不同催化剂对反应的影响,结果见表2.由表2可知:当体系中不含催化剂时,没有反应发生;当体系中存在摩尔分数0.67 %的金属酞菁催化剂时,通过液相色谱跟踪可知反应在2 h内即可完成,产率接近或超过文献报道的无取代铜酞菁催化剂(产率31.3 %,摩尔分数10 %催化剂,24 h)[12],并且催化剂用量更低、反应时间更短.此外,催化效果与金属中心、取代基位置和取代基大小也有一定的关系.如结构类似的铜酞菁催化剂比铁酞菁的催化效果略好(实验4与5).在α位具有取代基的铜酞菁催化剂比β位具有同样取代基的铜酞菁催化效果好(实验3与4);且α位取代基的空间位阻越大,催化效果越好(实验2与3).这是因为金属酞菁上取代基的存在,尤其是α位取代基的存在,可在一定程度上破坏酞菁的大平面分子结构,避免或减少聚合物的形成,进而增加轴向配位的位点,从而提高催化效率、缩短反应时间.

表2 金属酞菁配合物催化的咪唑N-芳基化反应
3 结 论
分别用钼酸铵催化法和DBU催化法合成了α位和β位取代的金属酞菁配合物,并通过多种表征手段证实了它们的分子结构.初步研究发现α位具有较大空间位阻的铜酞菁配合物在催化咪唑N-芳基化反应时具有较好的催化性能,并取得了比无取代铜酞菁配合物更好的催化效果.该研究结果为金属酞菁催化剂的设计奠定了一定的工作基础.
