Tubulin glutamylation: a skeleton key for neurodegenerative diseases
2019-07-18SiemvanderLaan,GeronimoDubra,KrzysztofRogowski
Microtubules (MTs) are cytoskeletal elements formed by a non-covalent association of α- and β-tubulin heterodimers. They provide structure and shape to all eukaryotic cells and are implicated in a variety of fundamental cellular processes including cell motility, cell division,mechanotransduction as well as long-distance intracellular cargo transport. In neurons, they constitute the molecular frame that maintains the lengthy axonal projections. In view of the relative size of some axons in the human body, which can reach up to 1 m, the active transport of e.g., vesicles over the MT arrays to the synaptic cleft, is of particular importance. Considering the numerous roles of MTs, it is not surprising that already 30 years ago, impairment of the MT-based system was proposed as a unifying hypothesis for the variable clinical presentations in Alzheimer's disease (Matsuyama and Jarvik, 1989). In this context,a key question is how the MT network accommodates all these different functions, often within the same cell? Current view is that every MT-dependent process is executed through the recruitment of a specific set of MT-associated proteins (MAPs) and molecular motors. Thus, it is of fundamental importance to understand how recruitment of these MAPs and motors is regulated. Since many of the MAPs and motors bind to the C-terminal tails of α- and β-tubulin, which are known to protrude from the MT surface, one important mechanism by which MTs may regulate the association of the effector proteins is through posttranslational modifications (PTMs).
The modifications that occur on the C-terminal tails consists of either addition or removal of amino acids including polyglutamylation,polyglycylation and detyrosination. Very recently we have identified the members of the vasohibin family as cysteine proteases responsible for tubulin detyrosination (Aillaud et al., 2017), a modification, which consists of proteolytic removal of the very C-terminal tyrosine residue present on α-tubulin. The reverse reaction that consists of reattachment of a tyrosine residue is carried out by an enzyme called tubulin tyrosine ligase (TTL). Moreover, the C-terminal tails of both α- and β-tubulin are also subjected to polymodifications namely polyglutamylation and polyglycylation. These modifications are reversible and consist of the enzymatic addition of sidechains composed of either glutamate or glycine to the gamma carboxyl groups of primary sequence glutamates. The enzymes, involved in the addition of both glutamylation and glycylation side chains, share a homology domain with TTL and thus are called tubulin tyrosine ligase like (TTLL). The human genome contains thirteen TTLL related genes. Nine of them are involved in tubulin polyglutamylation (TTLL1, TTLL2, TTLL4, TTLL5, TTLL6,TTLL7, TTLL9, TTLL11 and TTLL13) and three in tubulin polyglycylation (TTLL3, TTLL8 and TTLL10) (Rogowski et al., 2009), while one, TTLL12, remains without assigned function. On the other hand,tubulin deglutamylation has been shown to be catalyzed by a family of cytosolic carboxypeptidases (CCP), which is composed of six members(Rogowski et al., 2010). In contrast, the enzymes responsible for deglycylation remain to be discovered. Overall, this complex enzymatic machinery allows for spatial and temporal fine-tuning of the physicochemical properties of the MTs surface, ensuring functional diversification. In analogy to the “histone code”, this regulatory system was originally coined as the “tubulin code” in a seminal review (Verhey and Gaertig, 2007). A proof of concept of the “tubulin code” was provided in the context of in vitro studies showing that PTMs confer unique biochemical properties, drive dynein and kinesin motor velocity, processivity and the rates of MT depolymerisation (Sirajuddin et al., 2014).
While polyglycylation appears to be specific to cilia and flagella,polyglutamylation and detyrosination are more ubiquitous. Biochemical characterization of MTs obtained from brain tissue revealed the presence of extensive PTMs on the protruding C-terminal tails of αand β-tubulin with the most abundant modification being polyglutamylation. The first enzyme involved in glutamylation to be identified,TTLL1, was originally purified from mouse brain using classical biochemistry, and confirmed genetically by developing knockout Tetrahymena cells, which lacked homologous gene and showed reduced level of glutamylation (Janke et al., 2005). A comprehensive follow up study demonstrated that in humans, apart from TTLL1, eight additional members of the TTLL family encode tubulin glutamylases.These enzymes are characterized by different specificities with some of them preferentially being involved in initiation while the others in the elongation of the glutamate chain. The identification of the reverse enzymes, the CCPs, came with the analysis of Purkinje cell degeneration (pcd) mouse model. These mice exhibit ataxia, which results from postnatal degeneration of almost all Purkinje cells in the cerebellum.Genetic analysis revealed that pcd mice carry a mutation in the CCP1 gene, which encodes a protein having tubulin deglutamylase activity.As such, pcd mice display abnormally high level of polyglutamylation in the cerebellar neurons. Stunningly, the Purkinje cell degeneration phenotype observed in the pcd mice was rescued by a knockdown of TTLL1 glutamylase, demonstrating that neuronal death is indeed mediated by tubulin hyperglutamylation. These observations provided the first molecular link between altered levels of tubulin glutamylation and neurodegeneration (Rogowski et al., 2010).
Current view limits the regulation of tubulin glutamylation levels in the cells to direct competition between the forward and reverse enzymes and does not include additional regulators. Recently, we have identified cilia and spindle-associated protein (CSAP) as a master regulator of tubulin glutamylases (Bompard et al., 2018). We found that expression of CSAP enhances overall activity of all autonomously active glutamylating enzymes and in the case of TTLL5 and TTLL7 also potentiates their elongase activity. Moreover, biochemical analysis revealed that CSAP interacts with TTLL glutamylases and appears to regulate their protein abundance through stabilization (Figure 1A). In turn, due to its high affinity for MTs, CSAP redirects glutamylase activity from tubulin towards MTs. By exploring the human protein atlas(Uhlén et al., 2015), we found that CSAP has a striking distribution in human tissue and is preferentially expressed in brain (Figure 1B). Thus,we propose that neurons utilize the expression of CSAP, as a regulatory mechanism to increase glutamylase activity specifically on MTs. This strongly suggests that CSAP plays an important role in neuron biology,possibly by regulating MT stability and tubulin glutamylation levels.In this respect, it would be of interest to further study the function of CSAP in neurons especially that previous work demonstrated its importance in neuronal development using zebrafish as a model (Backer et al., 2012).
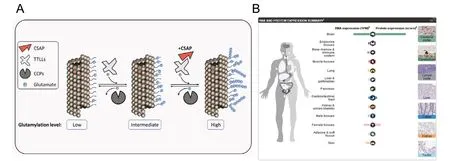
Figure 1 Cilia and spindle-associated protein (CSAP) function and tissue distribution in humans.(A) Schematic representation of the role of CSAP protein in glutamylation of microtubules. (B) Protein expression of CSAP in different organs of the human body. Data are obtained from Human Protein Atlas available from www.proteinatlas.org. TTLL: Tubulin tyrosine ligase like; CCP: cytosolic carboxypeptidases.
In the light of the functional importance of the levels of tubulin glutamylation in neuronal homeostasis, it is relevant to consider the potential mechanisms that might contribute to hyper-glutamylation. The best-documented mechanism so far involves inactivating mutations in the CCP deglutamylases, such as the ones described in the pcd mice.Similar outcome is expected for activating mutations in TTLL glutamylases, which have not yet been reported. Moreover, our latest discovery of CSAP as a potent activator of the forward enzymes opens up the possibility that changes in the expression of non-enzymatic regulators could also lead to abnormal accumulation of tubulin glutamylation and subsequent neurodegeneration.
A very recent study found that patients with infantile-onset progressive neurodegeneration carry inactivating mutations in the CCP1 gene,demonstrating the relevance of this mechanism in human disease (Shashi et al., 2018). Given the well-established role of tubulin glutamylation as an activator of MT severing enzymes, spastin and katanin appear as primary candidates responsible for neurodegeneration. Both severing enzymes have been shown to have the potential to progressively reduce MT mass through severing-mediated disassembly. However, genetic removal of spastin in pcd mice background did not seem to rescue cell death of the Purkinje neurons (Magiera et al., 2018). One explanation for the lack of rescue was provided by an elegant in vitro study, which showed that tubulin glutamylation increases spastin activity only within a certain range, while above a given threshold it becomes inhibitory.Thus, although spastin might be transiently activated, eventual increase in the length of glutamate sidechains is likely to ultimately become inhibitory. In the future, it will be important to further investigate the role of another severing enzyme, katanin, which also has been reported to be activated by tubulin glutamylation. However, in this case it remains to be established whether katanin activity also becomes inhibited by abnormally long glutamate chains. It is possible that contrary to spastin,activation of katanin increases linearly with the elongation of glutamate sidechains and results in progressive destruction of MT tracks.
Another potential mechanism responsible for hyperglutamylation dependent neurodegeneration is likely to be associated with the function of molecular motors. A study involving CCP1 deficient neurons reported a decrease in the overall transport of mitochondria, without affecting the speed or processivity. This suggest that tubulin tails modified by abnormally long glutamate chains, are likely to block the accessibility of the motors involved in the mitochondria transport to the MT tracks. The same mechanism might be responsible for the obstruction of the binding of structural MAPs such as, MAP1, MAP2 and Tau protein. In physiological conditions, these MAPs may interact with MTs without being affected by tubulin polyglutamylation. However,during neurodegeneration, the ever-increasing length of glutamate chains, which are eventually likely to become longer than the tubulin tails themselves, might interfere with the binding of structural MAPs.This could lead to decreased overall MT stability and progressive disassembly of MTs, ultimately resulting in the loss of neuronal polarity and death. Finally, considering the increasing number of the non-tubulin substrates of TTLLs and CCPs, the possibility that other proteins modified by glutamylation may also be involved in the molecular process of neurodegeneration, should not be ruled out.
Although multiple seemingly divergent mechanisms of neurodegeneration underlying Alzheimer's disease and many other neurodegenerative diseases have been described, we propose that a unifying primary event is microtubular dysfunction possibly due to altered PTMs. Our recent demonstration that vasohibin-1, a vascular-endothelial growth factor responsive gene, encodes tubulin detyrosinase opens the possibility that the enzymatic activity of this protein, which has been so far completely neglected, is important for vascularization (Aillaud et al., 2017). As such it is of interest to study the role of vasohibin-mediated detyrosination in neurovascular defects observed in Alzheimer's patients. Moreover,pathological increase in tau phosphorylation was shown to affect its interaction with MTs, a mechanism possibly contributing to neurodegeneration. The role of altered MT modifications in these neurodegenerative mechanisms needs to be thoroughly investigated.
Taken together, the latest advances in the field of tubulin PTMs, and more precisely glutamylation, clearly underline the importance of this mechanism in human health and disease. As such, research aimed at better understanding the molecular mechanism involved in neurodegeneration may further contribute to the identification of relevant targets. Accordingly, we propose that pharmacological inhibition of neuron specific glutamylases may represent a therapeutic avenue in the treatment of certain neurodegenerative diseases if not most.
This work was supported by ANR-13-JSV2-0002, Centre d'Excellence en maladies Neuro-dégénératives (CoEN) de Montpellier and France Alzheimer.
Siem van der Laan, Geronimo Dubra, Krzysztof Rogowski*
Institute of Human Genetics (IGH), Tubulin Code Team, Centre National de la Recherche Scientifique (CNRS)-University of Montpellier, Montpellier, France
*Correspondence to: Krzysztof Rogowski, PhD,krzysztof.rogowski@igh.cnrs.fr.
orcid: 0000-0001-8169-1963 (Siem van der Laan)0000-0002-0206-3289 (Krzysztof Rogowski)
Received: March 20, 2019
Accepted: April 25, 2019
doi: 10.4103/1673-5374.259611
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Hanin Abdel-Haq, National Center for Drug Research and Evaluation, Italy.
Additional file: Open peer review report 1.
杂志排行

中国神经再生研究(英文版)的其它文章
- Etomidate affects the anti-oxidant pathway to protect retinal ganglion cells after optic nerve transection
- Normal tension glaucoma: from the brain to the eye or the inverse?
- Mesenchymal stromal cell therapy for damaged retinal ganglion cells, is gold all that glitters?
- MicroRNAs as biomarkers of diabetic retinopathy and disease progression
- Diabetic neuropathy research: from mouse models to targets for treatment
- Potential therapeutic roles of retinoids for prevention of neuroinflammation and neurodegeneration in Alzheimer's disease