Potential therapeutic roles of retinoids for prevention of neuroinflammation and neurodegeneration in Alzheimer's disease
2019-07-18BhaskarDasSomsankarDasguptaSwapanRay
Bhaskar C. Das, Somsankar Dasgupta, Swapan K. Ray
1 Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, USA
2 Department of Neuroscience and Regenerative Medicine, Institute of Molecular Medicine and Genetics, Augusta University, Augusta, GA, USA
3 Department of Pathology, Microbiology, and Immunology, University of South Carolina School of Medicine, Columbia, SC, USA
Abstract All retinoids, which can be natural and synthetic, are chemically related to vitamin A. Both natural and synthetic retinoids use specific nuclear receptors such as retinoic acid receptors and retinoid X receptors to activate specific signaling pathways in the cells. Retinoic acid signaling is extremely important in the central nervous system. Impairment of retinoic acid signaling pathways causes severe pathological processes in the central nervous system, especially in the adult brain. Retinoids have major roles in neural patterning,differentiation, axon outgrowth in normal development, and function of the brain. Impaired retinoic acid signaling results in neuroinflammation, oxidative stress, mitochondrial malfunction, and neurodegeneration leading to progressive Alzheimer's disease, which is pathologically characterized by extra-neuronal accumulation of amyloid plaques (aggregated amyloid-beta) and intra-neurofibrillary tangles (hyperphosphorylated tau protein) in the temporal lobe of the brain. Alzheimer's disease is the most common cause of dementia and loss of memory in old adults. Inactive cholinergic neurotransmission is responsible for cognitive deficits in Alzheimer's disease patients. Deficiency or deprivation of retinoic acid in mice is associated with loss of spatial learning and memory. Retinoids inhibit expression of chemokines and neuroinflammatory cytokines in microglia and astrocytes, which are activated in Alzheimer's disease. Stimulation of retinoic acid receptors and retinoid X receptors slows down accumulation of amyloids, reduces neurodegeneration, and thereby prevents pathogenesis of Alzheimer's disease in mice. In this review, we described chemistry and biochemistry of some natural and synthetic retinoids and potentials of retinoids for prevention of neuroinflammation and neurodegeneration in Alzheimer's disease.
Key Words: Alzheimer's disease; amyloid plaques; neurofibrillary tangles; neuroinflammation; neurodegeneration;retinoids
Introduction
Retinoids are natural and synthetic derivatives of vitamin A. These compounds are remarkable in brain health and disease as they are known to play significant roles in the development and normal functions of the human brain (Das et al., 2014). So, there is a great interest in understanding the chemistry and biochemistry of the known and novel retinoids and their potential therapeutic applications to the treatment of brain diseases, especially Alzheimer's disease(AD). The vitamin A metabolite retinoic acid (RA) performs most of the physiological functions because RA has the ability of binding to the receptors of the nuclear receptor superfamily for regulation of expression of many genes in the cells (Lerner et al., 2012). Retinoids are highly regarded for their capability of modulating the expression of many genes that code for enzymes, neurotransmitter transporters, and receptors, transcription factors, cell surface receptors, and neuropeptide hormones (Goodman, 2006). Retinoids carry out transcription of their target genes through interaction with retinoid receptors such as retinoic acid receptors (RARα,β, and γ) and retinoid X receptors (RXRα, β, and γ), which themselves are transcriptional regulators and are known to be highly expressed in amygdala, prefrontal cortex, and hippocampal areas in the brain (Goodman and Pardee, 2003).Binding of nuclear receptors to a specific DNA site either repress or activate expression of target genes (Khorasanizadeh and Rastinejad, 2001). The functional responses of RA and their receptors are modulated by many co-activators and co-repressors (Jenster, 1998; Xu et al., 1999). Co-activators and co-repressors modify chromatin and/or interact with the typical transcriptional machinery for modulating transcription of the target genes (Lee et al., 2001).
Retinoid deficiency or mutation in RARβ and RXRγ genes is known to be associated with inhibition spatial learning and memory and also development of depression in animals.Studies showed that suppression of expression of RARα in rats, which were deprived of vitamin A, caused deposition of amyloid-beta (Aβ) peptide in the cerebral vessels (Shudo et al., 2009). Retinoids have important roles in prevention of neuroinflammatory responses for providing neuroprotection (Lee et al., 2009). Retinoids are known to down regulate expression of cytokines and inflammatory molecules in microglia (Goncalves et al., 2013). The agonists of retinoid receptors increase expression of choline acetyltransferase gene and vesicular acetylcholine transporter gene to enhance cholinergic neurotransmission (Mufson et al., 2008).
It is now widely known that elderly adults over age 65 are usually the AD patients. Aging is a major risk factor in developing AD. Currently, AD is the most common neurodegenerative disease that affect more than 15 million people worldwide (Andreeva et al., 2017). The demography of AD is rapidly expanding in the global populations. Clinical observations firmly show the association of AD with dementia and loss of memory. Neuropathologically, AD is characterized by extra-neuronal accumulation of amyloid plaques and intra-neuronal neurofibrillary tangles in temporal lobe of the brain. The amyloid plaques are composed of aggregated Aβ peptide while neurofibrillary tangles are hyperphosphorylated tau protein (Querfurth and LaFerla, 2010). Accumulation of these protein aggregates triggers neuroinflammation, oxidative stress, and mitochondrial damage leading to loss of not only neurons but also white matter in the brain.Emerging evidence suggests that AD pathology may result from a complex interplay between abnormal Aβ and tau proteins (Figure 1). According to the ‘amyloid hypothesis'of AD, accumulation of Aβ aggregates in the extracellular space of neurons in the brain is the primary cause for driving the pathogenesis for neurodegeneration and cognitive decline in AD patients (Hardy and Allsop, 1991; Musiek and Holtzman, 2015). The strength of amyloid hypothesis lies in its consistency with the genetic defects in AD, but it has deficiencies in explaining some important issues in AD.All attempts to develop drugs for targeting Aβ and treating AD have ended in failure (Karran and De Strooper, 2016).On the other hand, the ‘tau hypothesis' of AD states that hyper phosphorylation of tau protein is the main factor for formation of neurofibrillary tangles and progression of AD(Kametani and Hasegawa, 2018). The major weakness of the amyloid hypothesis is its inability in conclusively identifying the biochemical pathways that link amyloid plaque to tangle formation for neurodegeneration in AD (Götz et al., 2004;Eriksen and Janus, 2007). There are many other hypotheses about pathogenesis in AD and many drugs based on these hypotheses have been developed for treatment of AD (Du et al., 2018). Because AD is a multidimensional disease, it is now becoming clear that development of a drug with multiple therapeutic actions or combination of drugs with diverse activities for inhibition of pathogenesis will be required for successful treatment of AD.
In this review, we describe multiple therapeutic roles of natural and synthetic retinoids for prevention of neuroinflammation and neurodegeneration in AD. Promotion of novel and innovative research ideas on retinoids, hopefully,will enable the next generation of investigators in designing and synthesizing new multi-active retinoids to use as powerful therapeutic agents for prevention of pathogenesis in AD patients in the future.
Chemistry and Biochemistry of Natural and Synthetic Retinoids
It has long been known that retinoids play important roles in development and function of the embryonic and early postnatal brain (Jiang et al., 2012; Cunningham and Duester,2015; Bonney et al., 2018). However, increasing body of evidence indicates that retinoid signaling also plays important roles in the function of the adult brain (Lane and Bailey,2005; Kour and Rath, 2016; Mishra et al., 2018). Vitamin A(retinol) is the most multifunctional natural retinoid that regulates many biological processes such as embryonic development, cell differentiation, cell growth, and apoptosis as well as normal function of the brain (Khillan, 2014). Retinol is generally produced from pro-vitamin A carotenoids,which are supplied by many colorful fruits and vegetables or animal sources such as liver, egg yolks, or dairy products.Carotenoids are converted into vitamin A (retinol) in the small intestine of the animal body. Many photosynthetic plants, bacteria, and some fungi have the ability to biosynthesize pro-vitamin A carotenoids. But animals must consume carotenoids through their dietary sources for accumulation of vitamin A in the body (Weber and Grune, 2012;Green and Fascetti, 2016).
Retinoids are a group of compounds related to vitamin A,including its natural and synthetic derivatives, which have four isoprenoid units joined in a head-to-tail fashion. A retinoid has basic chemical structure that defines it a retinoid (Figure 2). Retinoids are unstable due to the presence of conjugated double bonds that easily undergo oxidation and/or isomerization in the presence of oxidants, light or excessive heat. Retinoids that contain alcoholic and carboxylic groups are soluble in methanol and ethanol, whereas the esterified long-chain fatty acid is only slightly soluble in alcohol but highly soluble in hexane. Epidemiological studies lend support to the hypothesis that higher dietary intake of pro-vitamin A carotenoids is associated with lower risk of many brain diseases, including AD (Li et al., 2012;Fiedor and Burda, 2014; Lakey-Beitia et al., 2017; Yang et al., 2017). Vitamin A also effectively increases visual tuning and prevents age-related macular degeneration (Cheung and Eaton, 2013; Harrison, 2019). The chemical structures of carotenoid precursors and natural retinoids contain a relatively long chain conjugated polyene (Figure 3). Carotenoids and retinoids are composed of conjugated polyene systems that absorb light in the visible and ultraviolet spectrums around 450 nm and within the rage of 325-380 nm, respectively(Furr, 2004). Colorimetric methods are commonly used for the estimation of carotenoids and retinoids.
For chemical synthesis of retinoids, many studies used the Wittig reaction to synthesize retinyl acetate and the ethyl ester of RA (Maercker, 1965). However, the Wittig reaction predominately synthesizes the cis isomer whereas trans olefin linkages are most frequently observed in the natural retinoids. Over the last few decades many modifications and alternatives to the Wittig olefination have been invented. In the Horner Wadsworth Emmons modification, replacement of the phosphonium salts with phosphonate esters produced trans or E-olefin. Two groups of investigators (Julia and Arnould, 1973; Koch and Gartner, 1997) employed similar olefination techniques in retinoid syntheses. Recently, researchers have shown that fully functional retinoid receptor agonists can be developed without the classic extended polyene chain. Toxicity profile and off-target binding of all-trans-retinoic acid (ATRA) and cis-RA (which are also metabolized by many cytoplasmic enzymes such as Cyp26,isomerases, and others) are problematic and thus their full potentials as novel pharmacological agents have not been well exploited. To overcome these problems, the synthetic chemistry community has developed many synthetic retinoids (Altucci et al., 2007; le Maire et al., 2012) using SAR(Structure Activity Relationship) analysis and computational modeling.
Many excellent reviews and research articles have been published highlighting several natural and synthetic retinoids in the drug discovery process and the signaling pathways (Kagechika and Shudo, 2005; Das et al., 2014;Haffez et al., 2018; Chisholm et al., 2019). Currently, there are several major synthetic retinoids that are being used as clinical agents (Figure 4). Clinically, isotretinoin (13-cis-RA) is the most effective drug for treatment of acne vulgaris although during the isotretinoin therapy some changes occur in hematological parameters that are within the normal range (Gencoglan et al., 2018). Isotretinoin therapy, which was well tolerated, improved sperm production in some men with infertility (Amory et al., 2017). Fenretinide or N-(4-hydroxyphenyl) retinamide is being developed as an anti-cancer drug in the clinics (Cooper et al., 2017). Bexarotene monotherapy, which is well-tolerated, is effective in cutaneous T-cell lymphomas (Hamada et al., 2017) and also it has shown efficacy in patients with peripheral T-cell lymphomas (Farhan et al., 2019). Multiple-dose administration of R667 (0.2-1 mg) for up to 16 days was well tolerated in patients with emphysema (Chiu et al., 2007). Tazarotene is a very effective treatment for plaque psoriasis, with significant decreases in plaque elevation and scaling following 12 weeks of therapy (Tanghetti et al., 2018). Its efficacy and tolerability can be further increased by combination with topical corticosteroids. A new topical formulation of combination of tazarotene and halobetasol seems to provide an optimal management strategy for plaque psoriasis (Tanghetti et al.,2018). Clinical trials for five years have eventually established that adapalene in its various pharmaceutical formulations is an important addition to the current treatments for acne vulgaris (Millikan, 2001). Our research group has been successful in synthesizing bororetinoids, some of which have been used in preclinical studies (Zhong et al., 2011).
Both natural and synthetic retinoids are being extensively studied for functional neuroprotection in the central nervous system (CNS) injuries and diseases (Mey, 2006;Chakrabarti et al., 2016). The process of biosynthesis of RA starts with either the conversion of retinol to retinal or with retinal itself (Figure 5). Retinol, also known as vitamin A,is obtained from food sources (Sommer and Vyas, 2012).RA must be biosynthesized from retinol by two oxidation steps. Retinol is converted into retinal through the action of retinol dehydrogenase and the transformation of NAD+to NADH (Lidén and Eriksson, 2006; Hong et al., 2015). From retinal, RA is biosynthesized in the next oxidation step. This biosynthetic reaction requires enzymes known as retinaldehyde dehydrogenases (RALHDs or ALDHs), with common enzyme types being ALDH1A1-3 and ALDH8 (Duester,2008; Kedishvili, 2016). In the cytoplasm, retinol is bound to cellular retinol-binding proteins (CRBPs) while RA is bound to cellular RA-binding proteins (CRABPs) (Napoli,2017). CRBPs are divided into two classes (CRBP type I and II); similarly, CRABPs are also broken down into two classes(CRABP type I and type II) and they carry RA to the RARs and RXRs that are similar but differ in amino acid sequences and ligand bindings. The carrier proteins CRBPs are involved in transport and metabolism of retinol while CRABPs are involved in the regulation of many RA signaling pathways and making RA available to the nuclear receptors (Zhang et al.,2012).
Extra-Neuronal Increase in Amyloid-Beta Oligomers and Accumulation of Amyloid-Beta Plagues Leading to Pathogenesis in Alzheimer's Disease
A certain quantity of the Aβ is necessary for transmission of information to the neurons in the brain. Emerging evidence shows both physiological and pathological functions of Aβ on various stages of the synaptic vesicle cycle, from post-fusion membrane recovery to trafficking, docking, and priming of synaptic vesicles for fusion and release of neurotransmitters (Ovsepian et al., 2018). The mechanisms of storage and release of neurotransmitters at axon terminals are summarized in the widely-known hypothesis of synaptic vesicle cycle (Südhof, 1995), which is also the primary site of Aβ production (Müller et al., 2017). Two findings, (i) neuronal activity triggers formation of Aβ and (ii) increase in Aβ causes decrease in excitatory synaptic transmission (Kamenetz et al., 2003), led to the hypothesis that Aβ normally serve as a negative feedback signaling pathway and increase in synaptic activity increases processing of amyloid precursor protein (APP) to Aβ, which reduces synaptic activity(Venkitaramani et al., 2007). A recent report suggests that different isoforms, concentrations, and aggregation status of Aβ may differently influence synaptic function and dysfunction (Gulisano et al., 2018). The inhibition of endogenous Aβ impairs synaptic plasticity and memory, strongly indicating that the peptide is essential for healthy function of the brain.However, an increase in oligomeric Aβ has been related to synaptic dysfunction, which is the earliest sign of pathogenesis in AD.
It is now widely known that the major histopathological hallmark of AD is the accumulation of amyloid plaques composed of aggregation of Aβ peptide in different areas of the brain (parenchyma of the amygdala, hippocampus,and neocortex) (Reiss et al., 2018). Proteolysis of APP may occur by the selective actions of α-, β- and γ-secretases in two distinct pathways (Figure 6). The non-amyloidogenic cleavage of APP occurs on the cell surface, while internalization of APP and its amyloidogenic processing results in Aβ production, release, and oligomerization. Aβ peptide, which may contain 36-43 amino acids, is produced from the APP.Aβ peptide is a portion of the transmembrane domain and the extracellular domain of the APP, which occurs as several isoforms of 695, 751, and 770 amino acids. The non-amyloidogenic (normal) pathway of APP proteolysis uses α-secretase to produce an N-terminal fragment called APPα and a membrane-bound C-terminal fragment α. The neuroprotective APPα may play roles enhancing synaptogenesis, neurite outgrowth, and neuronal survival. In the membrane, C-terminal fragment α is cleaved by γ-secretase to yield a soluble N-terminal fragment (p3) and a membrane-bound C-terminal fragment called APP intracellular domain (AICD).The functions of AICD may include nuclear signaling via transcriptional regulation and axonal transport through its ability to associate with different proteins. The amyloidogenic (abnormal) pathway of APP proteolysis employs β-secretase to produce a N-terminal fragment called APPβ and a membrane-bound C-terminal fragment β. Then, C-terminal fragment β is cleaved by γ-secretase to generate a soluble N-terminal fragment (Aβ) and the membrane-bound C-terminal fragment (AICD) as before. Notably, Aβ is required for neuronal function but its accumulation in the extracellular space causes its aggregation to form Aβ plaques in the brain. The deleterious effects of Aβ on neuronal and synaptic function eventually cause neurodegeneration in AD.
The understanding of the processing of APP in two different pathways and its metabolites is crucial for development of novel therapeutics for AD (O'Brien and Wong, 2011).Many recent studies are revealing not only the regulation of APP processing but also physiological as well as pathological functions of APP and its metabolites (Zhang et al.,2011, 2012). The non-amyloidogenic pathway uses α- and γ-secretases and produces the peptides to provide helpful neurotrophic effects (Allinson et al., 2003; Haass et al.,2012). In contrast, the amyloidogenic pathway employs βand γ-secretases and produces Aβ peptides (Sinha et al.,1999; Francis et al., 2002). The Aβ peptides produced are prone to accumulation and aggregation. The predominantly produced Aβ peptides via the amyloidogenic pathway are 40 residues in length (Aβ1-40 or Aβ40) and monomers of Aβ40 are nontoxic. Only a small percentage of Aβ peptides contain 42 residues in length (Aβ1-42 or Aβ42). Increased plasma levels of Aβ42 have been correlated with occurrence of AD(Mayeux et al. 1999). Because Aβ42 has extra two amino acids, it has a greater tendency of misfolding and aggregation that may lead to formation of the neurotoxic amyloid deposits contributing to pathogenesis of AD (Ahmed et al., 2010).Dysregulation in synthesis, processing, and clearance leading to accumulation of aggregated Aβ42 peptides was thought to be the starting point of AD. Although it has previously been suggested that an accumulation of Aβ42 plaques promote formation of neurofibrillary tangles and ultimately neuronal death, it is now evident that soluble rather than accumulated Aβ is related to dementia (Nimmrich and Ebert, 2009). Soluble Aβ oligomers specifically interfere with synaptic function and are associated with neuropathology in AD (Lesné et al., 2013). It is now known that Aβ oligomers do not induce neuronal death, but prolongation of synaptic dysfunction ultimately causes degeneration of synapses abolishing their ability to encode and retrieve memories in the AD brain. The implication of Aβ42 in synaptic dysfunction may provide a new target for therapeutic intervention in AD (Marsh and Alifragis, 2018).
Intra-Neuronal Formation of Neurofibrillary Tangles for Pathogenesis in Alzheimer's Disease
Neurofibrillary tangles are aggregates of hyperphosphorylated tau protein, which is now most widely considered to be the primary marker of AD (Kolarova et al., 2012; Sierra-Fonseca and Gosselink, 2018). However, it has been demonstrated that significant loss of neurons occurs before the formation of neurofibrillary tangles and that neurofibrillary tangles account for only a small amount (around 8%) of loss of neurons (Kril et al., 2002). Only an increase in neurofibrillary tangle load is associated with severity and chronic aggression in AD patients (Lai et al., 2010). In addition to supporting neuronal architecture, microtubules are known to track and efficiently transport nutrients, molecules, and information in the neurons (Dent and Baas, 2014). The fiber-like protein tau is responsible for maintaining the stability of microtubules(Feinstein and Wilson, 2005). The threads of tau proteins become tangled and twisted due to their hyperphosphorylation by kinases such as glycogen synthase kinase 3 and p70 S6 kinase, and thus microtubules become unstable and disintegrate, causing collapse of the entire neuron transport system in AD patients (Brion, 1998). Moreover, recent studies suggest that other post-translation modifications (glycosylation,glycation, prolyl-isomerization, cleavage or truncation,nitration, polyamination, ubiquitination, sumoylation, and oxidation) of tau protein (Martin et al., 2011) and also tau self-aggregation (Farías et al., 2011) significantly contribute to pathogenesis and neurodegeneration in AD.
Neuroinflammation and Neurodegeneration in Alzheimer's Disease
Two major neuropathological features of AD are the extracellular accumulation of Aβ peptide into amyloid plaques and the intraneuronal formation neurofibrillary tangles of hyperphosphorylated tau protein, both of which are known to contribute to neuroinflammation and neurodegeneration(Hung et al., 2016; Cai et al., 2018). Neuroinflammatory responses are highly responsible for pathogenesis in AD(Shadfar et al., 2015; Sawikr et al., 2017; Ahmad et al., 2019).Chronic neuroinflammation and impairment of lipid homeostasis can lead to pathogenesis and neurodegeneration in AD (Hampel, 2012). It has been reported that an anti-ceramide antibody increases amyloid plaque formation and serum exosomes in a mouse model of AD (Dinkins et al.,2015). In the brains of AD patients, activated microglia and astrocytes reside very near to the plaques suggesting that activation of glial cells may be related to the formation of Aβ plaques (Wyss-Coray and Mucke, 2002). Microglial cells exhibit a main role for recognition and elimination of Aβ42 oligomers by increasing their phagocytic activity (Zotova et al., 2011; Sha et al., 2014). However, a more recent study suggests that chronic infusion of Aβ42 oligomers induces sustained neuroinflammation and activates microglia in rat hippocampus leading to spatial memory decline, as seen in AD (Fekete et al., 2018).
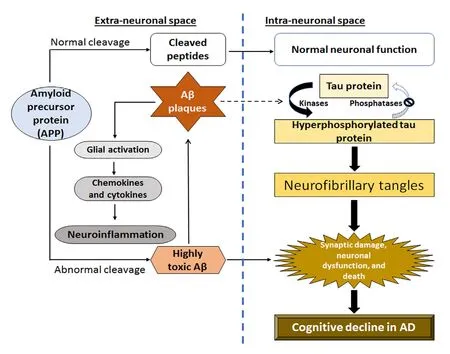
Figure 1 Schematic representation of a complex interplay between abnormal amyloid-beta (Aβ) and tau proteins leading to neuroinflammation and neurodegeneration in Alzheimer's disease(AD).Aβ aggregates into plaques outside the neurons, while abnormal tau proteins accumulate inside the neurons in specific regions of the brain involved in spatial learning and memory. After accumulation of Aβ plaques occurs significantly, the abnormal tau proteins spread rapidly throughout the brain leading to significant neuroinflammation,neurodegeneration, and cognitive deficits in AD.
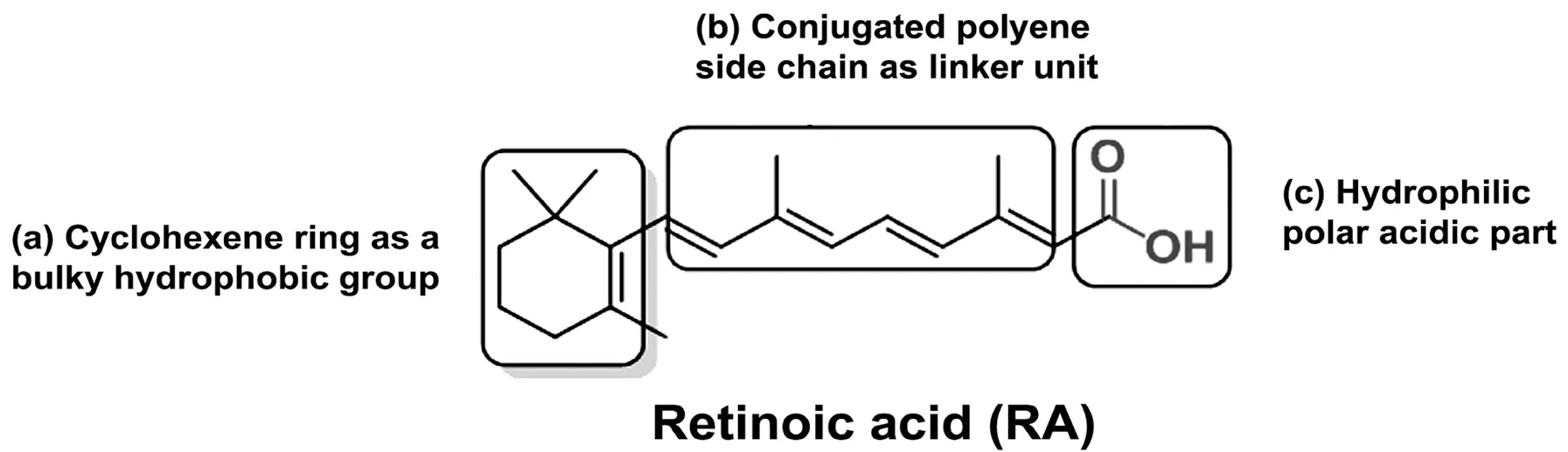
Figure 2 Basic chemical structure of retinoic acid (RA, a natural retinoid) with its different parts.The chemical structure of a retinoid has three parts: (a) a trimethylated cyclohexene ring that acts as a bulky hydrophobic group, (b) a conjugated tetraene side chain that serves as a linker unit, and (c) a typical carboxylic acid part that is hydrophilic with a polar carbon-oxygen functional group. The word ‘retinoid' includes only a few endogenous (naturally occurring) and a large group of synthetic derivatives of vitamin A.
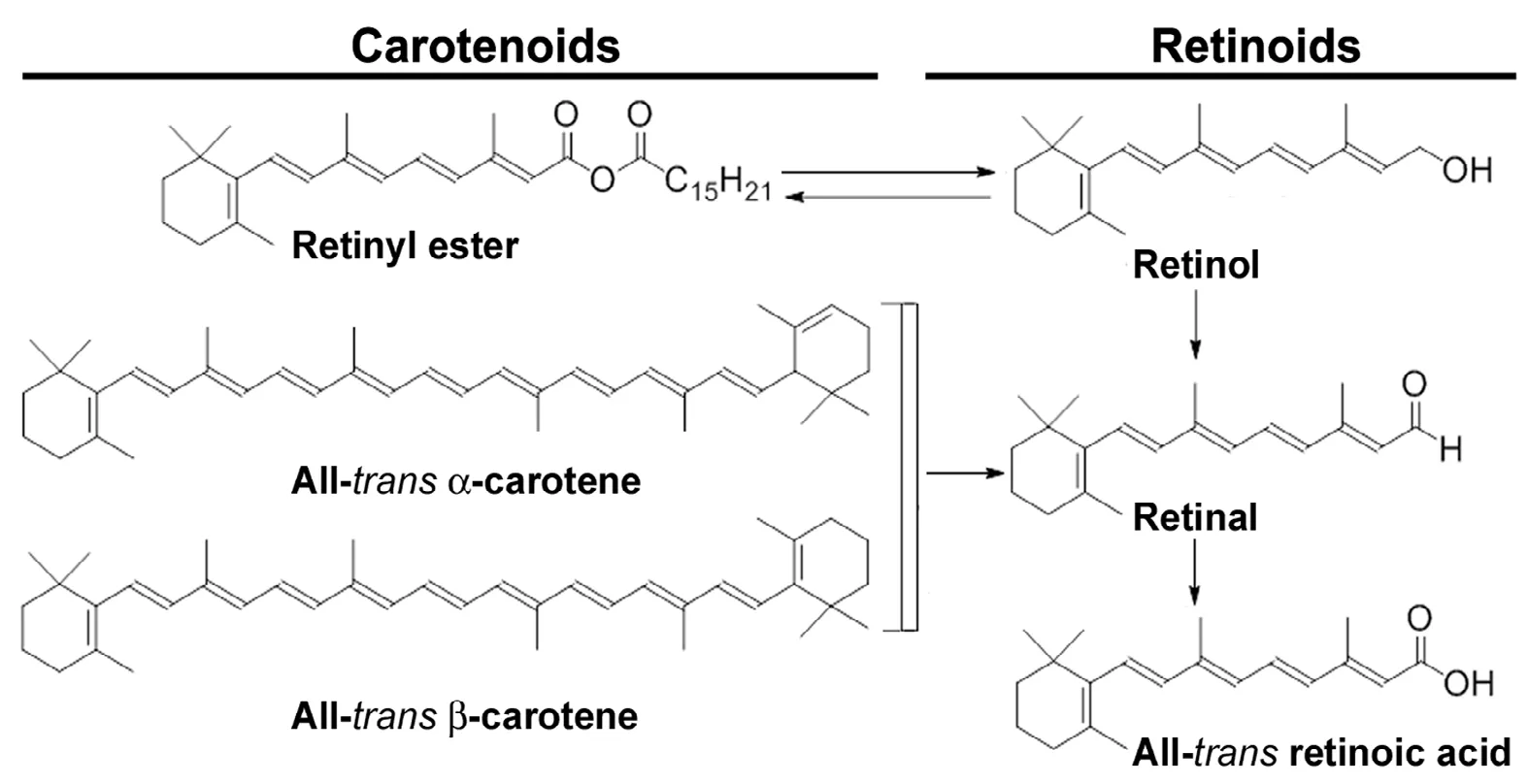
Figure 3 Chemical structures of natural carotenoids and retinoids.Carotenoids are precursors of vitamin A. Carotenoid precursors are found in plant-based foods such as dark and yellow vegetables, carrots, and fruits.Natural retinoids are chemical derivatives of vitamin A(retinol). Natural retinoids are found in animal-based foods such as liver, kidney, eggs, and dairy products.
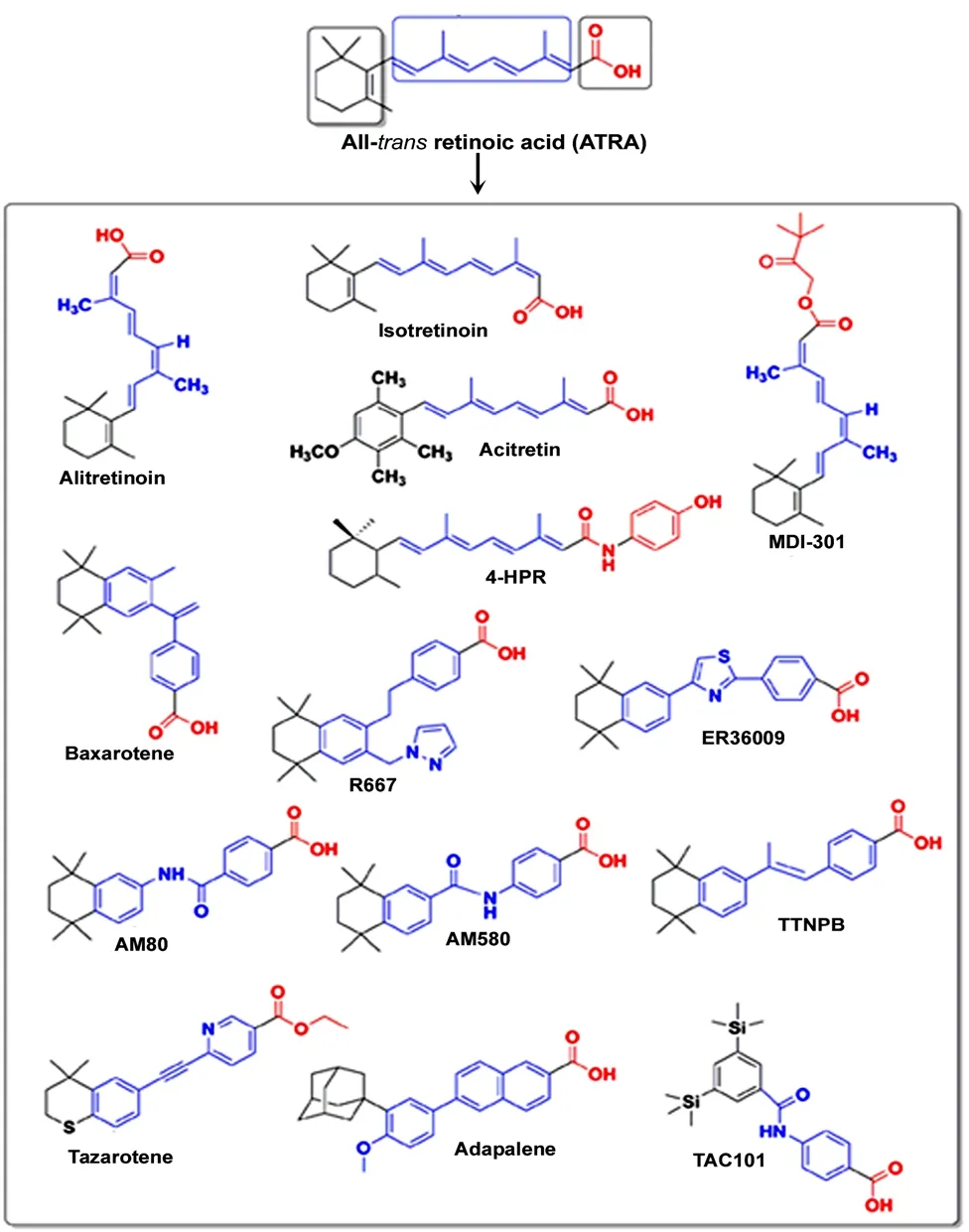
Figure 4 Chemical structures of some major synthetic retinoids used as clinical agents.Synthetic retinoids are produced from all-trans-retinoic acid (ATRA) with a goal of achieving high therapeutic effects and eliminating side effects in the clinics. The first orally administered synthetic retinoid to become approved for clinical use in the United States was 13-cis-retinoic acid(isotretinoin). Black showed the hydrophobic group, blue showed the linker unit, and red showed the polar carbon-oxygen functional group.
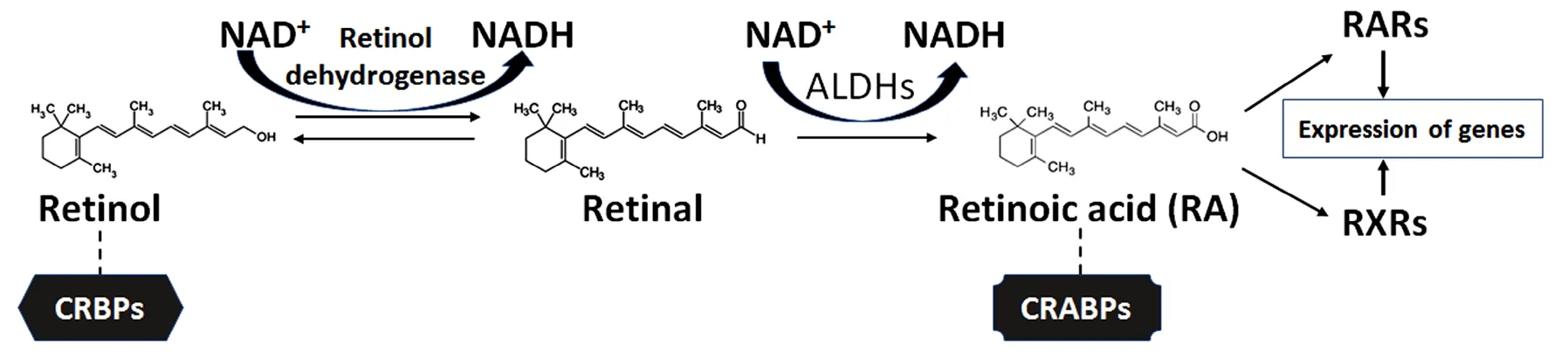
Figure 5 Vitamin A (retinol) as the starting compound for biosynthesis of retinoic acid (RA) in the cells.In the cytoplasm, retinol remains bound to cellular retinol-binding proteins (CRBPs) for its movement and role in metabolism, but RA remains bound to cellular RA-binding proteins (CRABPs) for its movement for regulating many signaling pathways. Also, RA with the help of CRABPs can move to the nuclear receptors (RARs and RXRs) for expression of specific genes. RARs: Retinoic acid receptors; RXRs: retinoid X receptors.
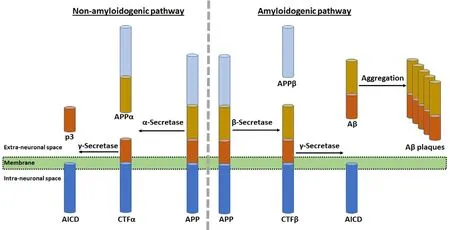
Figure 6 Amyloid precursor protein (APP)proteolysis in two different pathways and role of amyloid-beta (Aβ) plaques in neurodegeneration in the AD brain.The non-amyloidogenic (normal) pathway of APP proteolysis produces an N-terminal fragment (called APPα) and a membrane-bound C-terminal fragment α (CTFα).In the membrane, CTFα is then cleaved to yield a soluble N-terminal fragment (p3) and a membrane-bound C-terminal fragment called APP intracellular domain (AICD). The amyloidogenic (abnormal) pathway of APP proteolysis produces N-terminal fragment(called APPβ) and membrane-bound C-terminal fragment β (CTFβ). Then, CTFβ is cleaved to generate a soluble N-terminal fragment (Aβ)and membrane-bound C-terminal fragment(AICD) as before. Accumulation of Aβ in the extracellular space causes its aggregation to form Aβ plaques in the brain.
It has previously been demonstrated that chronic activation of microglia releases various chemokines and cytokines such as interleukin (IL)-1, IL-6, and tumor necrosis factor-alpha (Akiyama et al., 2000) and activates the complement system for catastrophic effects in inducing neuroinflammation and progression of AD (Holmes et al.,2009). Cholesterol has a role in pathogenesis in AD because high levels of serum cholesterol can significantly promote APP processing for Aβ metabolism leading to accumulation of Aβ plaques (Ledesma and Dotti, 2012). Alterations in expression of the genes, which are responsible for cholesterol homeostasis, are known to be important risk factors in pathogenesis of AD (Rogaeva et al., 2007). The interconnections between Aβ plaque and innate immune response are widely characterized and recent studies suggest the existence of interplays between tau pathology and the innate/adaptive immune responses (Laurent et al., 2018). Improper microglial function, due to either aberrant activation or decrease in phagocytic functionality, can occur during aging and in course of development of AD leading to neuroinflammation that ultimately contributes to neurodegeneration (Labzin et al., 2018). Early disease-provoking neuroinflammation could start decades before the presentation of severe cognitive impairments or dementia in the AD patients (Eikelenboom et al., 2010; Cuello, 2017). It is thought that early neuroinflammation and multiple dysfunctional pathways in the CNS can be promising therapeutic targets as we continue to search for a definite diagnosis of AD preclinical stages (Cuello, 2017;Elfakhri et al., 2019).
Currently Available Prescription Medications for Treatment of Alzheimer's Disease
There is no cure yet for AD. However, some prescription medications are currently available for AD patients for improvement of their cognitive and behavioral symptoms(Ghezzi et al., 2013; Kumar et al., 2015). Acetylcholinesterase inhibitors and glutamate receptor antagonists are approved by the US Food and Drug Administration (FDA) to treat AD and recently there have been some other advances in the pharmacotherapy for AD (Khoury et al., 2017). Depending on the clinical stages (early, moderate, and severe) of AD,four commonly prescribed medications are used (Figure 7).Donepezil (Aricept®) is approved by the FDA for all stages of AD (Lee et al., 2015; Kim et al., 2017). Rivastigmine (Exelon®)(Birks and Grimley Evans, 2015) and galantamine (Razadyne®) (Lilienfeld, 2002) are approved by the FDA for mild to moderate AD. Aricept®, Exelon®, and Razadyne®are acetylcholinesterase inhibitors that improve cognitive functions(learning and memory) (Rodda et al., 2009). But these medications show side effects such as nausea, vomiting, appetite loss, and increased rate of bowel movements (Kobayashi et al., 2016). Also, acetylcholinesterase inhibitors become less effective over time. Memantine (Namenda®) is the primary drug approved by the FDA for treatment of moderate to severe stages of AD (Kishi et al., 2018). Namenda®, which is an orally active glutamate receptor antagonist, can improve memory, attention, reason, language, and the ability to perform simple tasks (Matsunaga et al., 2015). But it causes side effects such as dizziness, headache, confusion, and constipation in AD patients (Perras, 2005). We described (Table 1)for the readers to quickly scan and extract information about the mostly used prescription medications for treatment of different stages of AD, their mechanisms of action, beneficial effects, and also adverse effects.
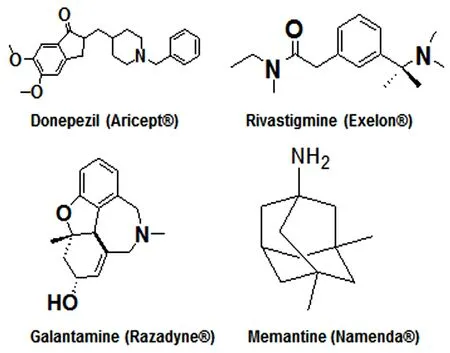
Figure 7 The drugs approved by the US Food and Drug Administration for treatment of Alzheimer's disease at different clinical stages.Aricept®, Exelon®, and Razadyne® are acetylcholinesterase inhibitors that block breakdown of the neurotransmitter acetylcholine by the enzyme acetylcholinesterase and thus they increase level and action of the neurotransmitter in the brain of Alzheimer's disease patients. Namenda® is a glutamate receptor antagonist that blocks the damaging effects of excessive amount of glutamate, also a neurotransmitter, produced in the brain of Alzheimer's disease patients.
The FDA-approved treatments offer neither promise of cure nor turnaround of the disease. These treatments, however, may alleviate some of the AD symptoms temporarily and thus help improve the health-related quality of life and somewhat delay placement of individual AD patients into the institutional care (Atri, 2011). All the current AD therapies, including acetylcholinesterase inhibitors and glutamate receptor antagonists, have limited scope and provide limited benefits in AD, which is a multifaced disease. No current therapy is yet capable of targeting the Aβ aggregation, which is crucial in pathogenesis of AD. New avenues need to be explored urgently to design and discover powerful drugs that possess the capability of targeting multiple pathological pathways to prevent pathogenesis of AD. Present treatment algorithms and potential future treatments for AD have been extensively described in a very recent review article (Grossberg et al., 2019).

Table 1 Currently available prescription drugs for treatment of different stages of Alzheimer's disease
Retinoids for Prevention of Neuroinflammation in Alzheimer's Disease
Neuroinflammation is an important player in the pathogenesis of AD (Regen et al., 2017). Inflammation causes synaptic dysfunction and neurodegeneration in the brain. Production of excessive neuroinflammatory mediators induces production and aggregation of Aβ peptide and hyperphosphorylated tau protein. Aggregates of Aβ peptide and hyperphosphorylated tau protein generate chemokines and cytokines.Intense neuroinflammatory responses are regularly detected in AD patients as well as in animal models of this disease(Johnston et al., 2011). Macrophages and microglia act as scavengers to remove Aβ aggregates via phagocytosis in the brain (Weitz and Town, 2012). But the presence of neuroinflammatory cytokines is known to block this phagocytosis(Weitz and Town, 2012). Optimal microglial function is necessary for scavenging tasks, but chronic activation of these cells in the brain also causes proinflammatory responses, oxidative stress, degradation of neuroprotective retinoids, and down regulation of RA signaling, promoting degeneration of surrounding healthy neurons (Regen et al., 2017).
During AD pathogenesis, Aβ-stimulated signaling pathways induce synthesis and release of proinflammatory cytokines (e.g., IL-1β, IL-6, tumor necrosis factor-alpha), chemokines (e.g., C-C motif chemokine ligand 2), and acute phase proteins as well as reactive nitrogen species and reactive oxygen species and can further promote plaque formation(Fiala, 2010). Astrogliosis, microgliosis, and chronic neuroinflammation are highly notable hallmarks in AD patients(Weisman et al., 2006). Aβ triggers production of proinflammatory cytokines and chemokines by the astrocytes and microglia but retinoids can suppress production of these proinflammatory mediators by interaction with RARs that are expressed in astrocytes and microglia (Shudo et al., 2009). It has been reported that retinoids cause activation of RAR and RXR to modulate functions of astrocytes and the microglia to reduce the production of proinflammatory cytokines and chemokines (Sodhi and Singh, 2014).
Prevention of neuroinflammatory responses is one of the most important goals in the treatment of AD. Roles of retinoids for prevention of neuroinflammation have been reported in neurodegenerative processes by earlier studies(Kuenzli et al., 2004). Since retinoids significantly inhibit generation of IL-6 (Zitnik et al., 1994; Kagechika et al., 1997),the strategy for down regulation of IL-6 by retinoids may be a useful therapy against AD. Retinoids have been observed to suppress lipopolysaccharide-induced or Aβ-induced tumor necrosis factor-alpha production and to inhibit expression of inducible nitric oxide synthase in activated microglia by inhibiting nuclear translocation of the nuclear factor-kappa B (Dheen et al., 2005; Kaur et al., 2006). More recent studies also show that ATRA inhibits lipopolysaccharide-induced neuroinflammation, amyloidogenesis, and memory deficits in old rats (Behairi et al., 2016) and promotes proliferation of neural stem cells along with suppression of activation of microglia leading to adult neurogenesis in the hippocampus in a mouse model of AD (Takamura et al., 2017).
Neuroinflammation inhibitory roles of an RAR agonist Am80 (Tamibarotene) was investigated in lipopolysaccharide-induced neuroinflammation model in vivo and the results demonstrated that Am80 could promote the production of brain-derived neurotrophic factor providing neuroprotection in pathological conditions (Katsuki et al., 2009).Am580 caused suppression of inflammatory cell death in cultured cortical neurons following exposure to Aβ (Jarvis et al., 2010). Retinoids play significant roles in inhibiting neuroinflammatory responses and promoting phagocytosis of Aβ aggregates in various neurodegenerative conditions including AD. Currently, intense research activities are underway for understanding the molecular mechanisms of action of retinoids and carotenoids for prevention of neuroinflammation in AD (Mohammadzadeh et al., 2017).
Because retinoids and carotenoids are potent anti-inflammatory and anti-oxidative agents, they provide neuroprotection. They are capable of suppression of AD progression through multiple mechanisms such as inhibition of production as well as accumulation of Aβ, suppression of oxidative stress, and inhibition of secretion of pro-inflammatory mediators so as to improve cognitive functions (Mohammadzadeh et al., 2017). All these recent studies strongly suggest that retinoids and carotenoids work through multiple pathways to provide potent neuroprotective effects in AD. A very recent report suggested that ATRA attenuated neuroinflammation by modulation of expression of Sirtuin 1, a class III histone deacetylase and a member of the Sirtuin family of proteins,and nuclear factor-kappa B in the rat brain (Priyanka et al.,2018).
Retinoids for Prevention of Neurodegeneration in Alzheimer's Disease
Dietary supplementation of carotenoids has been shown to play a crucial role in preventing several neurodegenerative diseases including AD (Obulesu et al., 2011). Retinoids are involved in neuronal patterning, differentiation, axonal outgrowth, and axonal regeneration (Maden, 2007; Puttagunta and Di Giovanni, 2012). Retinoid deprivation leads to impairment of normal brain development and function,resulting in the appearance of symptoms of different neurodegenerative diseases including AD (Etchamendy et al.,2003; Sánchez-Hernández et al., 2016; Zeng et al., 2017).Investigations indicate that retinoids can induce generation of specific neuronal cell types and further regenerate axons after damage (Maden, 2007). In addition, retinoids are involved in the maintenance of the differentiated state of adult neurons and neural stem cells as well as adequate levels of retinoid signaling for synaptic plasticity, learning,and memory in the adult brain (Lane and Bailey, 2005; Tafti and Ghyselinck, 2007). Thus, retinoids appear to be essential for the normal maintenance of the brain function and for the treatment of different neurodegenerative diseases of the brain, including AD (Dräger, 2006; Fukasawa et al., 2012;Niewiadomska-Cimicka et al., 2017).
Vitamin A and other retinoids can directly inhibit formation of Aβ plaques in vivo, indicating potential therapeutic roles of retinoids for neuroprotection and thus prevention of pathogenesis in AD (Lerner et al., 2012). It has been proposed that RA can significantly potentiate neurotransmitter functions of acetylcholine in cholinergic neurons in the brain(Szutowicz et al., 2015). Degeneration of cholinergic neurons contributes to deficits in cognitive and memory functions(Schliebs et al. 2011). The cholinotrophic properties of RA and its derivatives may justify their use in the treatment of AD (Sodhi and Singh, 2013; Szutowicz et al., 2015).An effective treatment of AD in a mouse model required co-activation of RARα and RARβ (with the agonist Am80 or Tamibarotene) and RXRs (with the pan agonist HX630)(Kawahara et al., 2014). This study reported that co-administration of Am80 (0.5 mg/kg) and HX630 (5 mg/kg) for 17 days significantly improved memory deficits in AβPP23 mice, but administration of either agent alone showed no therapeutic effect. However, these investigators did not report any potential side effects of combination treatment with RAR and RXR agonists.
Bexarotene, which is a RXR agonist and also known as rexinoid, is repurposed in a recent study for the treatment of AD in mouse models (Mariani et al., 2017). Bexarotene was dispersed in water and administered in AD mice by oral gavage at the dose of 100 mg/kg daily and treatment began at either 3.5 or 7.5 months of age and continued for 15 days. Bexarotene treatment improved memory, olfactory cross habituation, and neuron survival, while reduced plaque burden, astrogliosis, and expression of inflammatory genes. Collectively, bexarotene treatment decreaed neuron loss and increased markers of synaptic integrity leading to improved cognition in the mice of aggressive AD. But this study did not report any side effects of bexarotene treatment.Bexarotene is usually used as a selective rexinoid for cancer treatment as mentioned above but it is now emerging as a viable candidate for clinical trials in AD (Koster et al., 2017).Treatment of cancer patients with bexarotene at a dose up to 300 mg/m2per day raised triglycerides up to 2.5 times and more than half of the patients experience hypothyroidism(Marshall et al., 2015). Increases in triglyceride and cholesterol levels reverted to normal levels following cessation of therapy, and the triglyceride and cholesterol levels could be clinically manageable with antilipidemic therapy during treatment with bexarotene. Overall, the mechanism of action of bexarotene and similar rexinoids still remains controversial because they demonstrate distinct adverse side effects in humans that may show more detrimental effects in elderly AD patients if taken over prolonged periods (Koster et al.,2017).
Conclusion
Application of natural and synthetic retinoids and their receptor agonists are under investigation to regulate the on-going processes of stem cell turnover, cell plasticity, and tissue regeneration. Impaired retinoid signaling promotes AD pathology. Because retinoids are small molecules, they can readily enter the tissues and therefore constitute promising therapeutic candidates. Their application at lower dose or in combination with other neuroprotective drugs could minimize unwanted side toxicity in non-target tissues. Retinoids thus represent a novel therapeutic strategy for AD treatment as they can block multiple pathological conditions of this disease, including plaque formation, neuroinflammatory responses, and neurodegeneration in the brain. Although many therapeutic applications of retinoids have been well studied, it will be highly critical to synthesize receptor subtype and isotype specific retinoids (to reduce toxicity,off-target binding, and increase specificity). To address these issues, our research group is designing and synthesizing new bororetinoids as promising pharmacological agents for the treatment of AD and other neurodegenerative diseases.
Author contributions:Manuscript concept: BCD and SKR; literature search and initial manuscript preparation: BCD and SKR; critical revision and final approval of the manuscript: BCD, SD, and SKR.
Conflicts of interest: None of the authors in this article has conflict of interest to declare.
Financial support:The work was supported in part by an award from the Soy Health Research Program (SHRP, United Soybean Board, Chesterfield, MO, USA) (to SKR), a grant (SCIRF-2015-I-01)from South Carolina Spinal Cord Injury Research Fund (Columbia,SC, USA) (to SKR), and earlier R01 grants (CA-091460, and NS-057811) (to SKR) from the National Institutes of Health (Bethesda,MD, USA).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Etomidate affects the anti-oxidant pathway to protect retinal ganglion cells after optic nerve transection
- Normal tension glaucoma: from the brain to the eye or the inverse?
- Mesenchymal stromal cell therapy for damaged retinal ganglion cells, is gold all that glitters?
- MicroRNAs as biomarkers of diabetic retinopathy and disease progression
- Diabetic neuropathy research: from mouse models to targets for treatment
- Sigma-2 receptor as a potential therapeutic target for treating central nervous system disorders