超高效液相串联质谱检测茶叶及茶饮料中咖啡因含量
2019-07-10王惠芳吴建兵钱志荣陈祝军
秦 园,王惠芳,吴建兵,钱志荣,吴 梅,陈祝军
(张家港市疾病预防控制中心,江苏张家港 215600)
咖啡因(Caffeine)是从茶叶、咖啡果中提炼出来的一种黄嘌呤生物碱化合物[1],是一种中枢神经兴奋剂,适度地使用有去除疲劳、兴奋神经并恢复精力的作用,临床上常用于治疗昏迷复苏和神经衰弱[2-3]。长期或过量地食用含咖啡因的食物或饮品会刺激中枢神经[4]、影响睡眠[5],甚至引起婴幼儿早产[6-7]等;一但停用还会出现精神萎糜、浑身困乏等戒断现象,危害人体健康[8-10]。因此,对不同产品及饮料中咖啡因含量快速、准确的测定,有助于人们科学、适量地摄入相关产品。
目前用于检测咖啡因的方法有:薄层色谱法[11]、紫外分光光度法[12-14]、气相色谱-质谱联用法(Gas chromatography-mass spectrometry,GC-MS)[15]、高效液相色谱法(High performance lipid chromatography,HPLC)[16-20]及液相色谱串联质谱法(Lipid chromatography-tandem mass spectrometry,LC-MS)[21-22]。其中薄层色谱法步骤复杂、有毒溶剂用量多且为半定量法;紫外分光光度法灵敏度较低;液相色谱检测周期长;气相色谱-质谱联用法无法直接测定,具有假阳性;液相串联质谱检测咖啡因的方法国内已有报道,陈彩云等[2]探究了液相串联质谱法检测巴西绿蜂胶制品中的咖啡因,而该方法用于检测茶及茶饮料中咖啡因含量报道较少,且专属性不够强。
本文探究超高效液相串联质谱法检测咖啡因的具体实验条件,并用于多种茶叶及茶饮料中咖啡因含量检测。对茶叶中咖啡因含量的检测方法进行补充,为证明该方法检测食品中咖啡因含量的可行性提供实验依据。
1 材料与方法
1.1 材料与仪器
咖啡因标准品(GBW(E)082413,浓度为0.50 mg/mL) 北京海岸鸿蒙标准物质技术有限责任公司;甲醇、乙腈 色谱纯,TEDIA公司;甲酸 色谱纯,上海国药集团化学试剂有限公司;去离子水 由去离子水器制得,美国Millipore公司;龙井 产地为浙江;碧螺春 产地为苏州;普洱 产地为云南;金骏眉、铁观音、大红袍 产地为福建;各茶饮料 均从市场自购。
UPLC/XEVO TQD液相色谱-串联质谱仪、ACQUITY UPLC BEH C18色谱柱(2.1 mm×50 mm,1.7 μm)、Waters 2695液相色谱仪、SunFireTMC18色谱柱(2.1 mm×250 mm,5 μm) 美国Waters公司;一次性针头过滤器(13 mm×0.22 μm) 上海安谱实验科技股份有限公司;调节式电炉 上海华联环境试验设备公司。
1.2 实验方法
1.2.1 样品前处理 茶叶:直接称取茶叶样品0.500 g(精确至0.001 g),置于250 mL锥形瓶,加入100 mL水,沸水浴30 min,不时振摇,取出锥形瓶流水冷却1 min,转移至250 mL容量瓶中,加水定容,摇匀,静置,取上清液经0.22 μm微孔滤膜过滤后,备用。
茶饮料:称取1.00 g(精确至0.001 g)样品,加水定容至2 mL,摇匀,经0.22 μm微孔滤膜过滤后,备用[23]。
1.2.2 标准溶液 配制吸取咖啡因标准溶液200 μL于100 mL容量瓶中,用4%甲醇水溶液稀释并定容,配制成1000 μg/L标准储备液,4 ℃避光保存,备用。
1.2.3 色谱条件 ACQUITY UPLC BEH C18色谱柱(2.1 mm×50 mm,1.7 μm)美国Waters公司;柱温为25 ℃;流动相:0.1%甲酸水溶液-乙腈(80∶20,v/v);流速为0.3 mL/min;分离时间5 min;进样体积10 μL。
1.2.4 质谱条件 电喷雾电离离子源(ESI);扫描方式为正离子扫描;MRM监测模式;脱溶剂气(N2)温度:500 ℃;毛细管电压:3.5KV;脱溶剂气(N2)流速:700L/Hr;锥孔电压:55V;碰撞气为高纯氩气。
1.2.5 方法学考察
1.2.5.1 准确度实验 方法准确度以加标回收率来判定,精确取出0.5 mL含已知咖啡因含量的金骏眉茶汤9份,分成3组,每组3份,于每组中加入0.5 mL咖啡因标准溶液,浓度分别为100、200、400 μg/L,按“1.2.3”色谱条件、“1.2.4”项质谱条件进行检测,记录峰面积,计算回收率。
1.2.5.2 精密度实验 “1.2.5.1”项中的各加标样品,按“1.2.3”色谱条件、“1.2.4”项质谱条件连续重复进样6次,记录峰面积,计算RSD。
1.3 数据处理
使用Excel软件,计算实验数据。SPSS 18.0软件进行统计学分析,茶叶及茶饮料样本结果以配对t检验进行统计分析,p<0.05为差异有统计学意义。
2 结果与分析
2.1 色谱条件建立
GB5009.139-2014《食品安全国家标准食品中咖啡因的测定》是检测食品中咖啡因现行有效的方法,本实验将该方法[23]中流动相条件:甲醇-纯水(24∶76,v/v),分别应用于Waters 2695液相色谱仪和超高液相串联质谱测定咖啡因标准溶液,其他色谱条件均参考GB5009.139-2014[23]。研究发现:甲醇-纯水(24∶76,v/v)流动相可在液相色谱仪上很好的分离出咖啡因,基线稳定,对目标峰响应无影响(见图1A),目标峰分离时间稳定为10.21 min;但该流动相用于超高效液相串联质谱检测咖啡因时,发现目标峰虽分离时间缩短(分离时间1.35 min),但其响应值易受基线干扰(见图1B)且加标回收率不理想,说明甲醇-纯水(24∶76,v/v)流动相条件不适用于超高效液相串联质谱测定咖啡因。
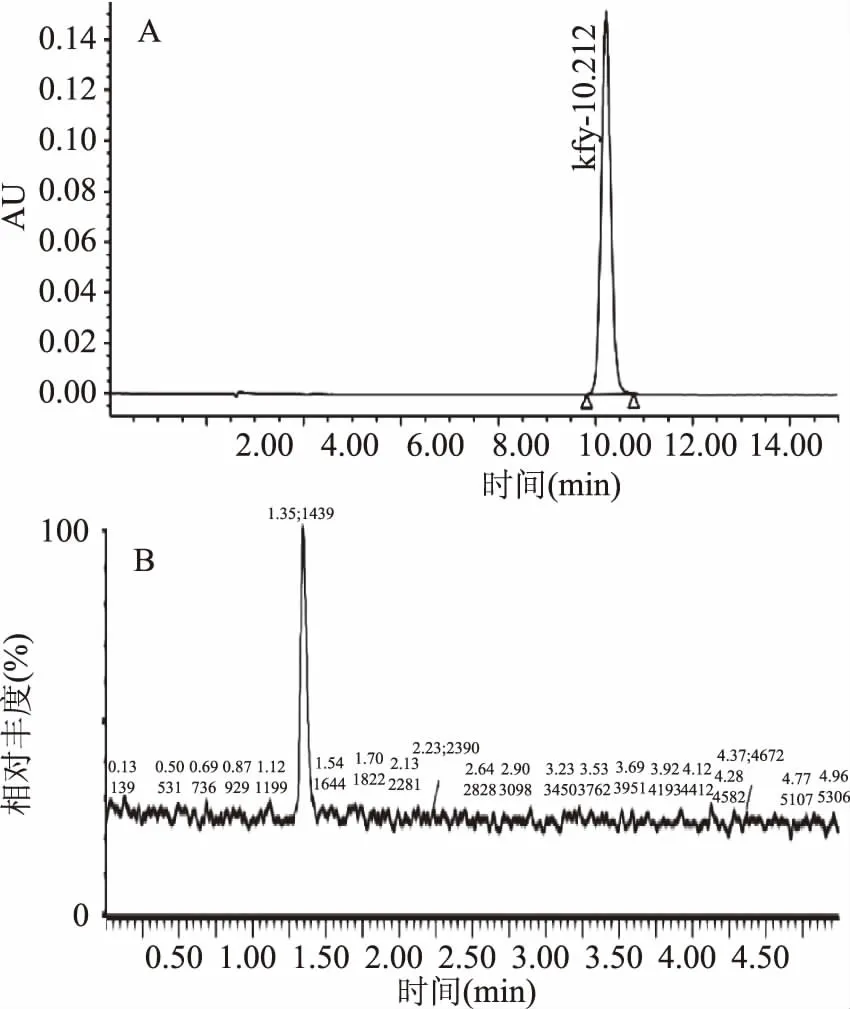
图1 UPLC-MS、LC检测的咖啡因标准品色谱图Fig.1 Chromatogram of standard caffeine detected by UPLC-MS and LC注:A.液相色谱仪检测咖啡因标准液; B.超高效液相串联质谱检测咖啡因标准液。
本文参考相关文献[2,24-25]关于咖啡因的检测,筛选出0.1%甲酸水溶液-乙腈(80∶20,v/v)流动相进行等度洗脱用于超高液相串联质谱检测咖啡因,实验结果显示:0.66 min目标峰即出现,响应值高、峰形好且不受基底干扰,能够准确分离和定量咖啡因(见图2),优于国标法[23]6.263 min出峰时间,因此将0.1%甲酸水溶液-乙腈(80∶20,v/v)流动相作为后续实验条件。
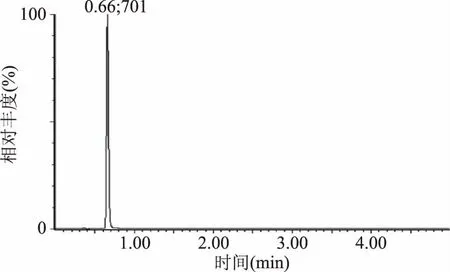
图2 UPLC-MS检测的咖啡因标准品多反应监测色谱图Fig.2 MRM chromatogram of standard caffeine detected by UPLC-MS
2.2 质谱条件优化
根据咖啡因的分子结构,本实验采用正离子(ESI+)作为离子化模式。质谱条件优化采用自动进样方式,将浓度为500 μg/L的咖啡因标准液以10 μL/min的速度注入质谱系统,在ESI+模式下,通过Q1全扫描找出母离子。实验结果显示,咖啡因在ESI+模式下有较高的响应值。同时考查脱溶剂气(N2)温度、毛细管电压、脱溶剂气(N2)流速、锥孔电压对离子强度的影响,并利用仪器自动优化目标化合物的碰撞能,最终选出母离子、两个峰度较大的特征性子离子和最优碰撞能参数,详见表1。

表1 咖啡因MRM参数Table 1 Parameters of Caffeine in multiple reaction monitoring(MRM)
2.3 方法学验证
2.3.1 检出限与标准曲线 以1000 μg/L咖啡因标准液为初始溶液,通过倍比稀释法,用纯水分别稀释成2、5、50、500、1000 μg/L的工作液,经0.22 μm微孔滤膜过滤,进样检测,以目标峰响应值为三倍信噪比(S/N=3)时相对应的工作液浓度作为该方法的检出限。实验结果显示:本方法中咖啡因的检出限为2.0 μg/L,优于咖啡因国标法[23]70 μg/L的检出限。
以咖啡因浓度分别为2、5、50、500、1000 μg/L的工作液进行实验。以溶液浓度(x,μg/L)为横坐标,各浓度对应的色谱峰面积(y)为纵坐标,进行线性回归计算,绘制标准曲线(见图3)。线性方程:y=40.3738x-18.1993,线性相关系数(r):0.9969,浓度范围在2.0~1000 μg/L之间线性关系良好。
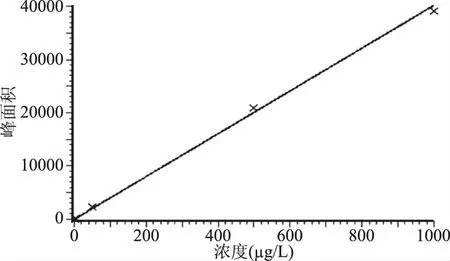
图3 咖啡因标准曲线Fig.3 Standard curve of caffeine
2.3.2 准确度和精密度 实验计算结果显示:不同标准液添加水平下,茶样品中咖啡因的回收率为90.2%~103.9%,表明该方法测定咖啡因含量的准确度良好;不同浓度标准液添加水平下,茶样品中咖啡因含量的相对标准偏差为1.00%~3.00%,表明该方法测定咖啡因含量的精密度良好。各水平加标回收率和相对标准偏差见表2。
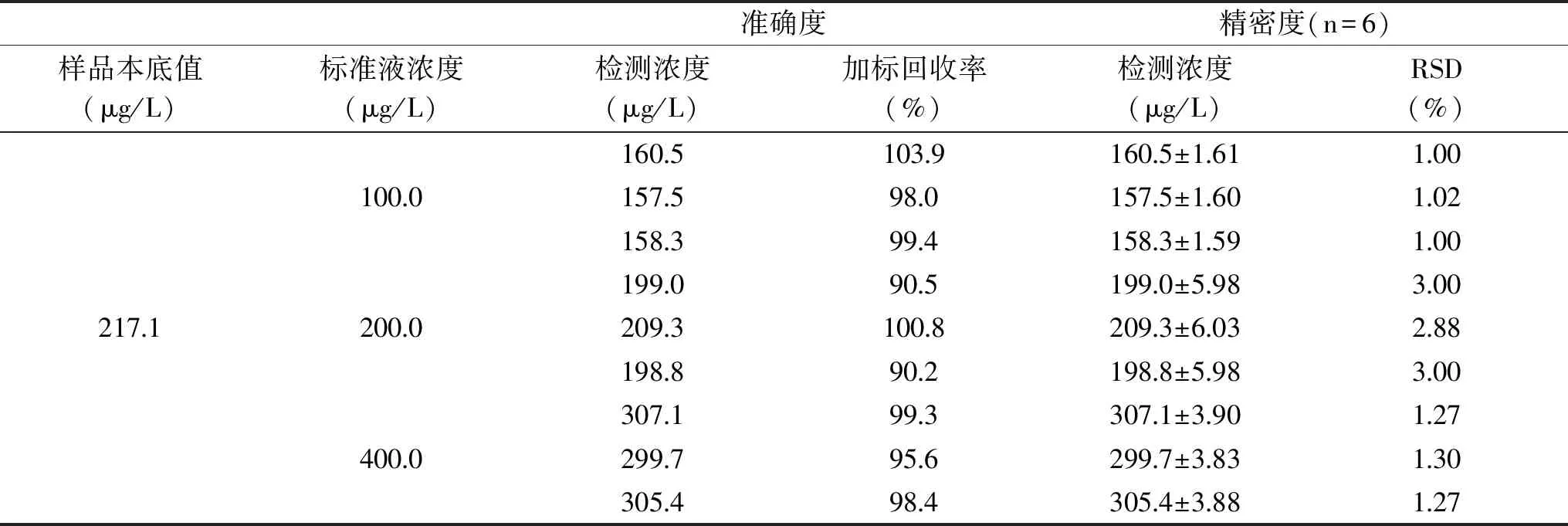
表2 UPLC-MS法检测咖啡因加标回收率和RSD测定结果Table 2 Precision and recoveries of UPLC-MS examining caffeine
2.4 实际样品检测
以本实验建立的超高效液相串联质谱法以及国标液相色谱法对绿茶、红茶、乌龙茶茶叶及4种茶饮料中的咖啡因含量进行测定,每个样品重复3次。实验结果显示:茶叶与茶饮料中均有咖啡因检出,茶叶中咖啡因含量在4.38~13.31 mg/g,茶饮料中咖啡因含量在0.07~0.16 mg/g,茶叶较茶饮料中咖啡因的含量高;红茶中咖啡因含量略低于绿茶、乌龙茶茶叶中咖啡因含量(见表3),这可能与不同茶叶的产地、制作方式等有关。
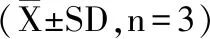
表3 两种方法测定茶叶及茶饮料中咖啡因含量Table 3 Detect caffeine in different tea and tea drinks with two
两种检测方法所得咖啡因含量结果经配对t检验统计分析(t=-0.820;p=0.433>0.05),无显著性差异;且UPLC-MS法及国标法检测咖啡因含量RSD分别为:2.05%~3.75%、1.18%~3.19%(表3),经配对t检验统计分析(t=1.897;p=0.09>0.05),无显著性差异;表明超高效液相串联质谱法检测咖啡因含量准确性可靠。
3 结论与讨论
本研究结果显示:超高液相串联质谱法检测咖啡因的检出时间为0.66 min;检出限为2.0 μg/L;线性浓度范围:2.0~1000 μg/L,相关系数(r)为0.9969;样品加标回收率达90.2%~103.9%;相对标准偏差在1.00%~3.00%之间(n=6)。与国标法[23]相比,本法具有检测时间短、灵敏度高等特点。
本文建立了超高液相串联质谱法对不同茶叶及茶饮料中的咖啡因含量测定的方法。与国标色谱法及现有其他检测方法相比[11-22,24-29],其检测速度、灵敏度均具有明显优势;且准确度、精密度、回收率均符合食品检测相关要求[30],可用于实际样品的检测;且该法对茶叶及茶饮料中咖啡因含量检测结果及RSD与国标法相比无统计学差异,结果准确可靠。该方法为实际情况下快速检测样品中咖啡因含量提供了高效、可靠的新方法。
