油酸改性Fe2(MoO4)3用于稠油水热催化降黏的研究
2019-07-08陆江银云庆庆
成 浪,李 玲,陆江银,云庆庆
(1.新疆大学石油天然气精细化工教育部重点实验室,乌鲁木齐 830046;2.新疆油田分公司实验检测研究院)
由于常规轻油资源的枯竭,在过去几十年稠油已成为主要研究热点[1]。然而,稠油黏度高、成分复杂,以经济有效的方式开采、输送稠油依然具有难度[2-3]。目前,循环蒸汽吞吐技术是最普遍且最有效的稠油开采技术,该技术是通过水蒸气给油藏提供热量从而达到稠油降黏目的[4-5]。Shokrlu等发现过热蒸汽不仅可以降低稠油的黏度,而且还能与稠油发生反应,改变稠油的性质与组成,并将此化学反应描述为水热裂解[6]。且在催化剂加入的情况下,水热裂解反应的效率可以得到很大程度上的提高,稠油降黏效果更加显著[7]。稠油水热催化改质降黏可实现稠油不可逆降黏,该技术具有潜在的应用前景,是未来经济高效的稠油开采技术[8]。
本研究用油酸对Fe2(MoO4)3进行改性,并用于新疆克拉玛依稠油的降黏,考察催化剂用量、反应温度、反应时间对稠油催化改质降黏的影响,通过稠油族组成分析、FT-IR、元素分析、1H NMR等分析手段对稠油降黏原因进行探讨。
1 实 验
1.1 主要试剂与设备
油酸、钼酸铵、三氯化铁、氢氧化钠、四氢萘、正庚烷、正己烷、甲苯、乙醇、石油醚,分析纯;三氧化二铝(层析用,100~200目);稠油(来自新疆克拉玛依,黏度(50 ℃)为13 200 mPa·s)。
CJF-0.2 L高温高压反应釜,NDJ-8S旋转黏度计,Vario MACRO cube 元素分析仪,EQUINOX 50红外光谱仪,BENFEN 3420气相色谱仪,Bruker D8 advance X射线粉末衍射仪,Varian INOVA-400超导核磁共振波谱仪,SDT Q600 热重分析仪。
1.2 催化剂OA-Fe2(MoO4)3的制备
将一定量的油酸(OA)与NaOH加入到500 mL单口烧瓶中,然后加入混合溶剂(蒸馏水、乙醇和正己烷体积比为1∶1∶4),升温至60 ℃恒温0.5 h。加入一定量的FeCl3·6H2O和(NH4)6Mo7O24·4H2O溶液,恒温4 h后,用分液漏斗滤掉水层,有机层用蒸馏水清洗后转入200 mL水热釜中,在160 ℃下保持8 h,然后产物用乙醇及蒸馏水清洗,最后在65 ℃下干燥12 h得到催化剂,记为OA-Fe2(MoO4)3。
1.3 稠油催化改质降黏试验
取50 g稠油置于高温高压反应釜中,加入一定量的水、供氢剂、催化剂后密封。通入N2,排出釜内空气后将反应压力升至3 MPa,将温度升至反应温度。反应一定时间后,停止反应。待反应釜冷却至室温后,取出油样,在50 ℃下(下同)测定降黏稠油的黏度,分析稠油族组成变化情况。稠油降黏率计算式如下:
式中:η为稠油降黏率;μ0为稠油初始黏度;μ为稠油水热催化降黏后的黏度。
1.4 表征与测试
族组成分析采用NB/SH/T 0509—2010方法;采用气相色谱仪分析裂解气组成;采用FT-IR分析自制催化剂、反应前后稠油及重组分的结构;采用元素分析仪分析稠油及重组分的C,H,N,S元素含量;采用XRD表征催化剂晶体结构;采用热重方法分析催化剂的热稳定性;采用1H NRM表征稠油及重组分的分子结构。
2 结果与讨论
2.1 催化剂表征
2.1.1 FT-IR图1为油酸和OA-Fe2(MoO4)3的FT-IR图谱。从图1可以看出:波数2 920 cm-1和2 851 cm-1处为甲基和亚甲基的吸收峰;波数744 cm-1处为MoO4八面体υ1、υ2的偶合,波数864 cm-1处为Mo—O键振动吸收峰[9],波数540 cm-1处为Fe—O键吸收峰;波数1 690 cm-1处的吸收峰表明催化剂含有C=O键;波数1 439 cm-1处是—COO—对称拉伸吸收峰,表明催化剂中包含了—COO—基团[10-11]。因此,可以认为Fe2(MoO4)3与油酸存在强烈作用并形成复合物,是所预想的催化剂。
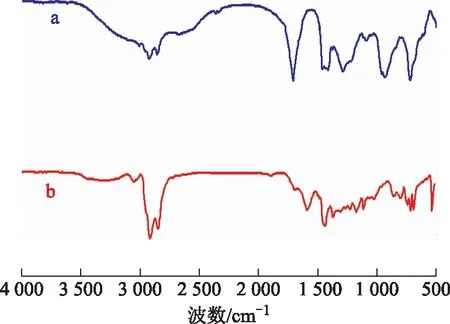
图1 油酸和OA-Fe2(MoO4)3的FT-IR图谱a—油酸; b—OA-Fe2(MoO4)3
2.1.2 XRD图2为OA-Fe2(MoO4)3样品的XRD图谱。从图2可以看出,OA-Fe2(MoO4)3样品的XRD图谱的特征衍射峰与3种Fe2(MoO4)3标准图谱吻合度较高,说明所制备的催化剂是Fe2(MoO4)3。图2中OA-Fe2(MoO4)3各特征衍射峰较为尖锐,杂峰较少,说明该催化剂纯度高且相对结晶度高。
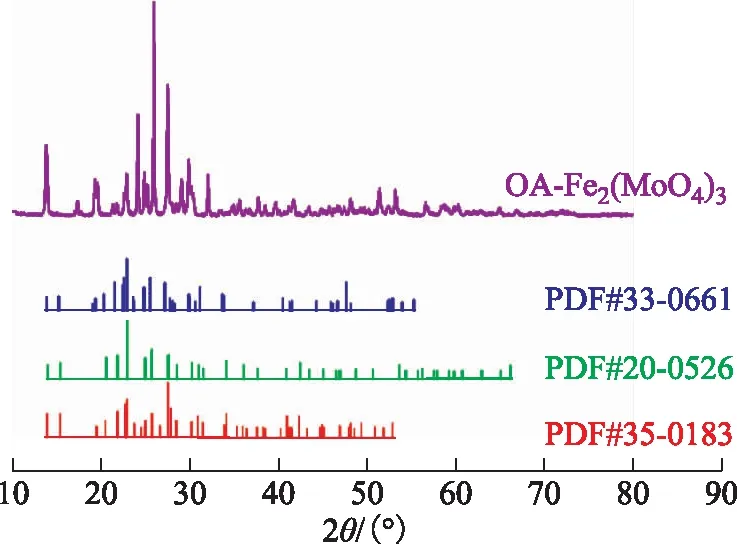
图2 OA-Fe2(MoO4)3的XRD图谱
2.1.3 热重分析为了更好地了解所制备的催化剂的热稳定性,对催化剂进行热重分析,结果见图3。样品的热重分析是从25 ℃到700 ℃,升温速率为10 ℃/min,N2气氛。从图3可以看出,OA-Fe2(MoO4)3的失重曲线大致可以分成3个阶段:第1阶段25~250 ℃的失重可以归因于结合水与有机溶剂的蒸发;第2阶段250~430 ℃的失重可能是由于油酸不饱和碳链断裂所致;第3阶段430~593 ℃的失重可以认为是剩余油酸的蒸发。在593 ℃时,Fe2(MoO4)3的残存质量约为83.31%。热重分析结果表明制备的催化剂有良好的热稳定性,可以用于稠油水热催化改质降黏。
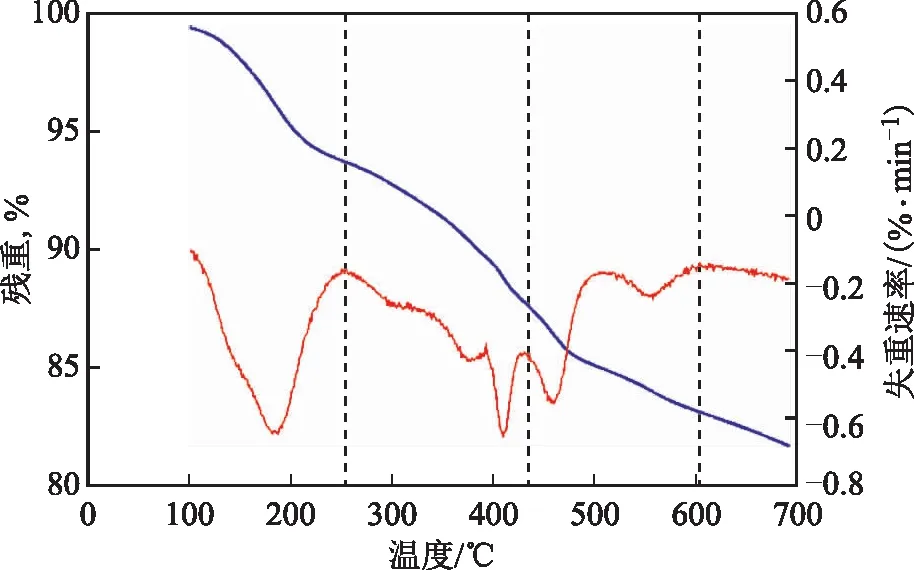
图3 OA-Fe2(MoO4)3的热重曲线 —残重; —失重速率
2.2 催化剂和反应条件对稠油降黏的影响
2.2.1 催化剂的影响将50 g稠油和1 mL四氢萘加入到反应釜中,反应温度控制在200 ℃,反应时间为12 h,考察催化剂的加入对稠油水热催化改质降黏效果的影响,结果见图4。其中,空白试验油样指不添加催化剂、水和四氢萘;水热裂解油样指仅添加水和四氢萘,其中油水质量比为7∶3;催化裂解油样指仅添加催化剂及四氢萘,其中催化剂添加量(w,下同)为0.2%;水热催化裂解油样指添加了水、催化剂和四氢萘,其中油水质量比为7∶3,催化剂添加量为0.2%。
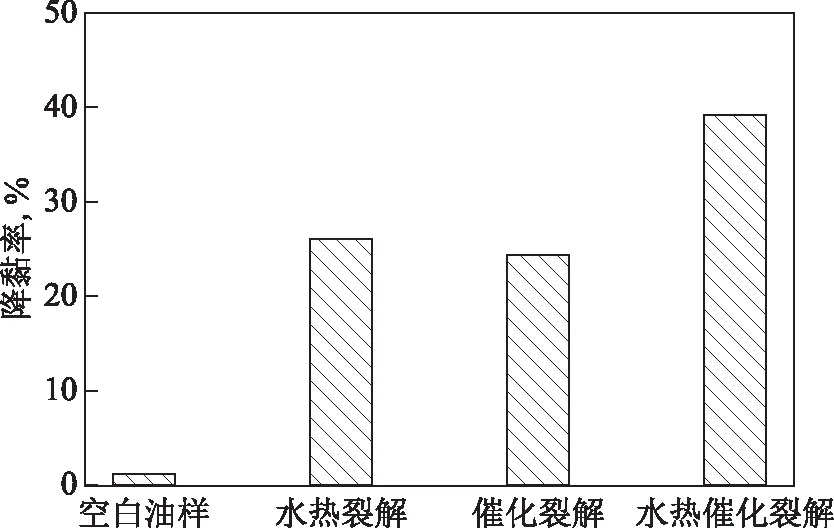
图4 催化剂对稠油水热裂解降黏效果影响
从图4可以看出:水热裂解、催化裂解对稠油均有降黏效果,降黏率分别为26.06%和24.30%;水热催化裂解后的降黏效果最好,降黏率达到39.24%,表明催化剂的加入有助于改善稠油水热裂解降黏效果,说明水与催化剂之间存在协同作用,两者共同作用更有助于稠油降黏。
2.2.2 催化剂添加量的影响将50 g稠油、水(油水质量比为7∶3)、1 mL四氢萘和催化剂加入到反应釜中,反应温度控制在200 ℃,反应时间为12 h,考察催化剂添加量对稠油水热催化降黏效果的影响,结果见图5。从图5可以看出:随催化剂添加量的增加,降黏效果随之提高;催化剂添加量小于0.4%时,降黏效果变化趋势明显,当催化剂添加量为0.4%时,稠油黏度降至7 440 mPa·s,降黏率为43.63%;当添加量继续增加至1.0%时,降黏效果变化趋势较为平缓,此时稠油黏度降至7 060 mPa·s,降黏率为46.51%,与催化剂添加量为0.4%时相比仅变化了2.88百分点。因此,催化剂最佳添加量选择0.4%。
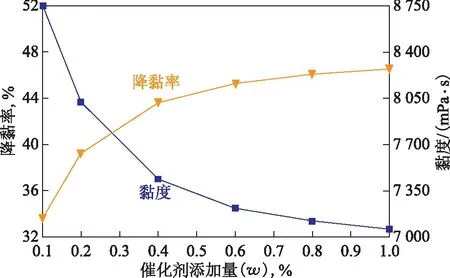
图5 催化剂添加量对稠油水热催化降黏效果的影响
2.2.3 反应温度的影响水热催化改质降黏过程中,需要满足一定的能量要求才能使稠油分子中部分化学键发生断裂[12]。因此,反应温度是反应过程中必须考察的重要指标。将50 g稠油、水(油水质量比为7∶3)、1 mL四氢萘和质量分数0.4%的催化剂加入到反应釜中,反应时间为12 h,考察反应温度对稠油水热催化降黏效果的影响,结果见图6。
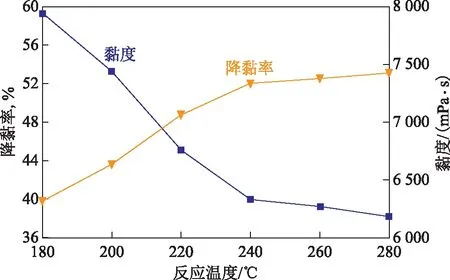
图6 反应温度对稠油水热催化降黏效果的影响
从图6可以看出:反应温度对降黏率有着重要的影响,随反应温度的升高,稠油黏度逐渐降低,降黏率逐渐增加;当反应温度为180~240 ℃时,稠油黏度从7 940 mPa·s降至6 430 mPa·s,降黏率从39.84%升至52.05%,说明该温度区间是催化剂发挥催化性能的最佳温度段;当温度区间为240~280 ℃时,黏度变化缓慢,降黏率趋于平稳。考虑到油藏以及水蒸气温度,最佳反应温度选择为240 ℃。
2.2.4 反应时间的影响将50 g稠油、水(油水质量比为7∶3)、1 mL四氢萘和质量分数0.4%的催化剂加入到反应釜中,反应温度控制在240 ℃,考察反应时间对水热催化改质后稠油黏度的影响,结果见图7。从图7可以看出:反应时间在12~36 h区间,稠油黏度随反应时间的增加而下降;在36 h之后,黏度下降趋势减缓并趋于稳定;当反应时间为36 h时,稠油黏度降至4 900 mPa·s,此时降黏率为61.21%。反应时间超过36 h后,黏度降低幅度较小的原因可能是水热催化改质过程中,胶质、沥青质是主要的反应产物,其含量随反应的进行逐步减少,反应速率也随之降低[13],此时可认为改质降黏反应基本完成,故最佳反应时间选择为36 h。

图7 反应时间对稠油水热催化降黏效果的影响
3 稠油水热催化改质降黏原因分析
3.1 油样族组成
表1为水热催化改质降黏在最佳反应条件下(催化剂添加量为0.4%,反应温度为240 ℃,反应时间为36 h)油样与空白试验油样族组成变化情况。从表1可以看出:水热催化改质后族组成发生了显著变化;胶质和沥青质质量分数分别从31.0%和13.2%降至20.7%和10.3%,分别降低10.3百分点和2.9百分点;饱和分和芳香分质量分数分别从32.9%和22.9%增至41.5%和27.5%,分别增加8.6百分点和4.6百分点。这表明水热催化反应可以有效促进稠油中的重组分(胶质和沥青质)裂解成轻组分(饱和分和芳香分)。
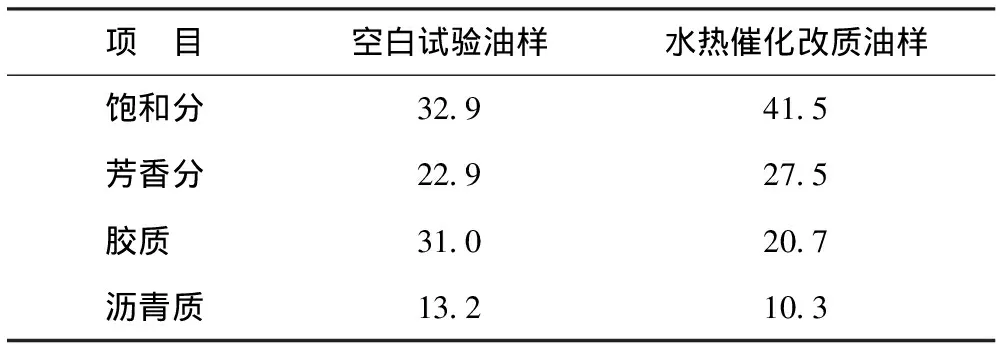
表1 空白试验油样与水热催化改质油样的族组成 w,%
3.2 裂解气分析
对空白试验和水热催化改质后的气相产物用气相色谱仪进行成分及含量分析,结果见表2。从表2可以看出,反应裂解气中主要含有CO、CO2、H2以及小分子烃类物质。其中,水热催化改质后CO2含量增加明显,说明稠油在水热催化改质过程中发生了脱羧反应;而烷烃、烯烃等物质的含量增加,说明催化裂解过程中稠油中重组分发生了解聚和桥键断裂,产生了小分子物质[14]。此外,C3和C4等组分含量增加则可能是自由基机理与碳正离子机理同样适用于本反应体系,稠油在催化剂的作用下,大分子发生脱侧链反应,进一步生成气态烃等小分子[15]。
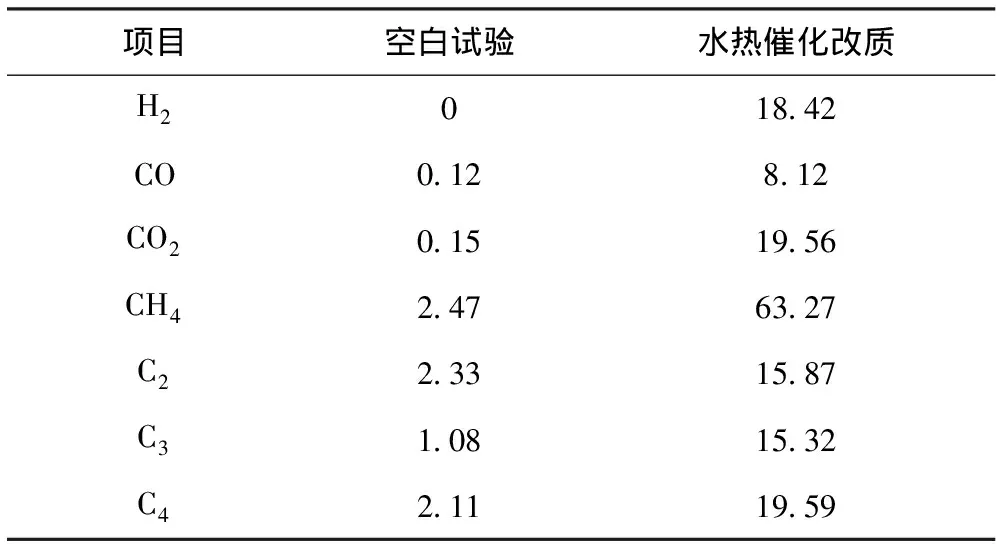
表2 空白试验与水热催化改质裂解气组成 φ,%
注:表中数据以N2体积分数为100%基础计算得到。
3.3 元素分析
表3为空白试验油样和水热催化改质油样的元素分析结果。从表3可以看出,油样的N、S质量分数分别从改质前的0.59%和1.01%降至改质后的0.58%和0.83%,C、H质量分数分别从改质前的87.37%和11.03%变为改质后的86.88%和11.71%,H/C原子比从改质前的1.51增至改质后的1.62。通过比较可知,水热改质后油样的N、S质量分数分别降低0.01百分点和0.18 百分点,C质量分数降低0.49百分点,H质量分数增加0.68百分点,H/C原子比增加了0.11。水热催化改质降黏后的油样H/C原子比略增加的原因可能是稠油中不饱和组分加氢的结果。而且,不难发现,与空白试验油样相比,水热催化改质后油样中的N、S含量均有一定程度降低,但S含量降低程度更大,说明水热催化裂解过程中C—S键、C—N键均发生了断裂,同时也说明C—S键比C—N键更易断裂。
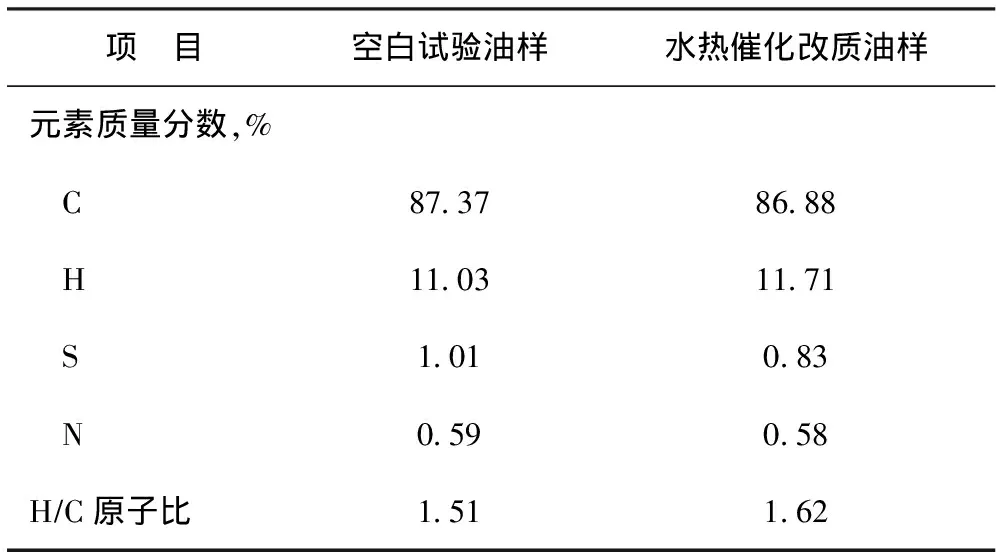
表3 空白试验油样与水热催化改质油样的元素含量
表4为空白试验油样与水热催化改质油样的重组分(胶质、沥青质)元素分析结果。从表4可以看出,水热催化改质后,胶质、沥青质中的S、N含量均有所降低,H/C原子比均有所增加。其中S含量的降低进一步说明了在水热催化改质过程中,C—S键确实发生了断裂,解释了稠油黏度降低的原因。两个重组分H/C原子比均增加,表明反应过程中加氢反应是同时进行的。
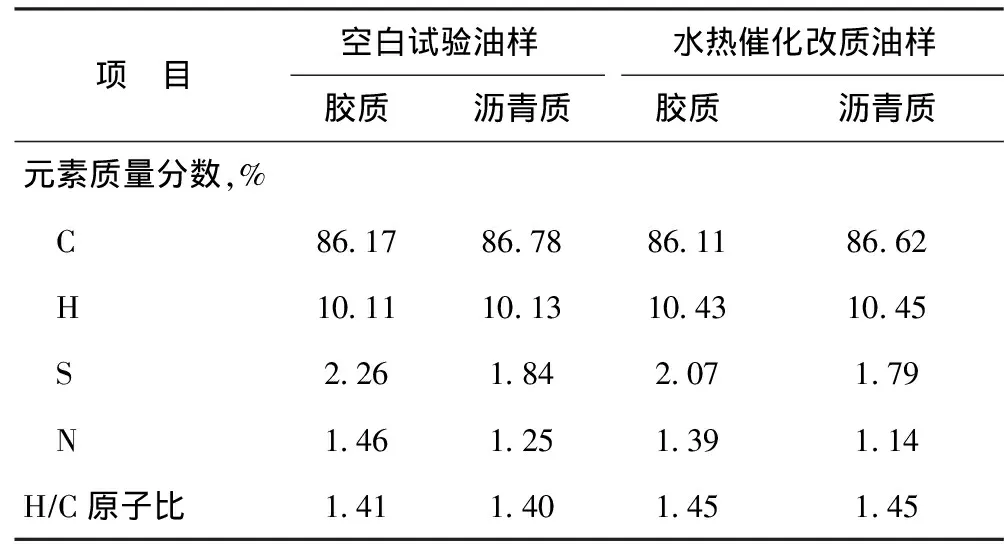
表4 空白试验油样与水热催化改质油样的胶质、沥青质的元素含量
3.4 稠油、胶质和沥青质的FT-IR分析
3.4.1 稠油的FT-IR分析图8为空白试验油样与水热催化改质油样的FT-IR图谱。从图8可以看出:波数2 922 cm-1和2 853 cm-1处分别是甲基、亚甲基伸缩振动特征吸收峰;波数1 603 cm-1处的吸收峰为稠环芳烃特征峰,水热催化改质后此峰增强,说明油样中多环芳烃数减少较为明显;波数1 454 cm-1和1 377 cm-1处分别是甲基、亚甲基弯曲振动特征吸收峰;波数742 cm-1处为长链烷烃(CH2)n(n≥4)特征吸收峰,水热催化改质后此峰增强,说明反应产生了更多的长链烷烃,且烃类的饱和度增大。
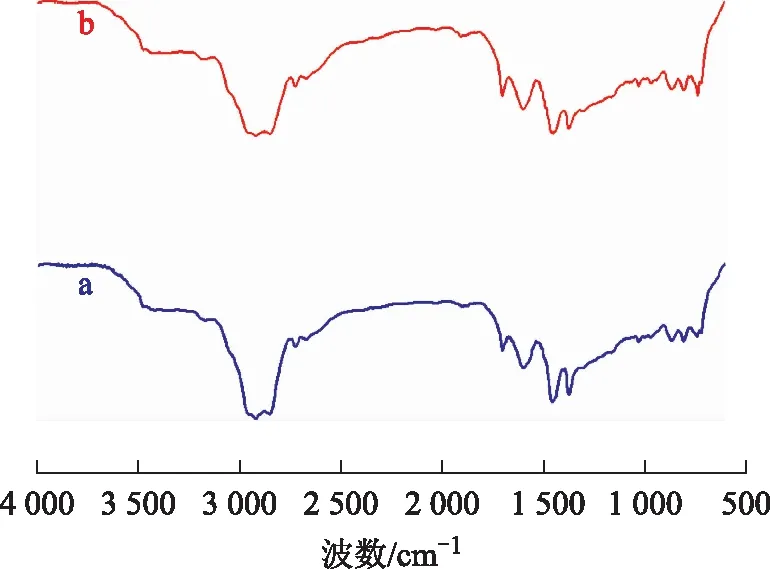
图8 空白试验油样和水热催化改质油样的FT-IR图谱a—空白试验油样; b—水热催化改质油样
3.4.2 胶质的FT-IR分析图9为空白试验油样与水热催化改质油样的胶质的FT-IR图谱。从图9可以看出:波数2 920 cm-1和2 851 cm-1处是甲基、亚甲基伸缩振动吸收峰;波数1 454 cm-1和1 373 cm-1处是甲基、亚甲基弯曲振动吸收峰;波数1 678 cm-1处是胶质中的C=O吸收峰,水热催化改质后此峰变弱,表明反应过程中发生了如C=O裂解的脱羧反应;波数1 597,1 373,864,812 cm-1处是胶质中的稠环芳烃吸收峰,水热催化改质后峰面积变小,说明在反应过程中发生了开环反应,芳香环数减少;波数1 234,1 180,1 026 cm-1处的峰在改质反应后减弱,表明有C—S,C—O,C—N键断裂。
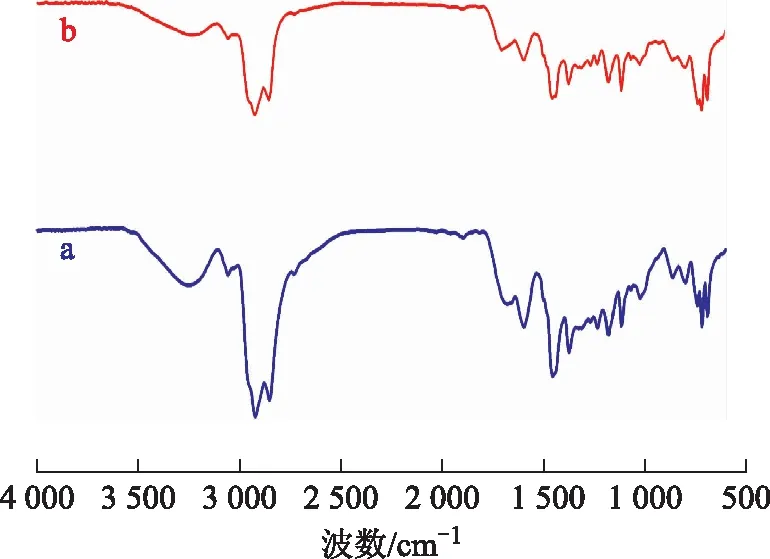
图9 空白试验油样和水热催化改质油样胶质的FT-IR图谱a—空白试验油样胶质; b—水热催化改质油样胶质
3.4.3 沥青质的FT-IR分析图10为空白试验油样与水热催化改质油样的沥青质的FT-IR图谱。从图10可以看出:波数2 920 cm-1和2 851 cm-1处是甲基、亚甲基的特征吸收峰;波数1 504 cm-1处的峰在水热催化改质后消失,说明存在部分脱烷基化反应[16];波数1 030,1 180,1 231 cm-1处的峰在水热催化改质后变弱,表明有C—N,C—O,C—S键断裂;波数802 cm-1处的峰在水热催化改质后变弱,说明有脱烷基链反应发生;波数1 593 cm-1处的峰在水热催化改质后增强,表明水热催化裂解过程中油样的芳香环数降低。
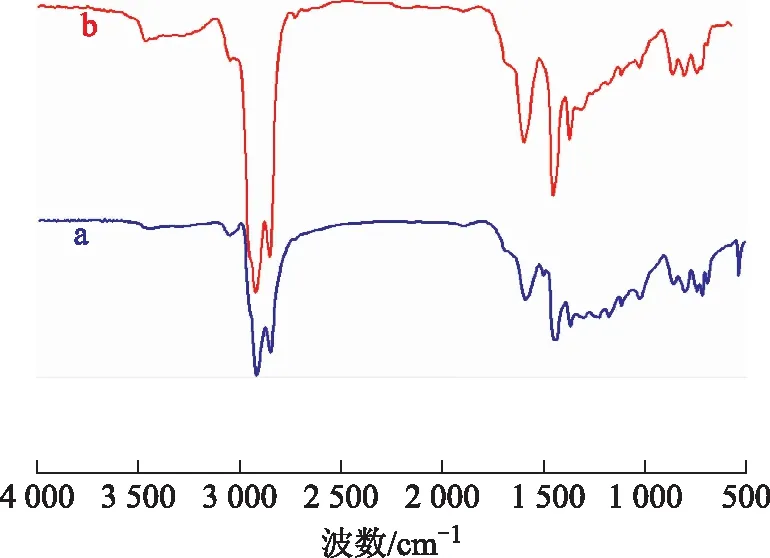
图10 空白试验油样和水热催化改质后油样沥青质的FT-IR图谱a—空白试验油样沥青质; b—水热催化改质油样沥青质
胶质、沥青质的FT-IR谱图说明其主要是由稠环芳烃、烷基链以及杂原子构成,所有变化情况都说明在水热催化改质降黏后,C—N,C—O,C—S键均有不同程度的断裂,这一结果与元素分析结果一致。
3.5 胶质与沥青质的1H NMR分析
油样的胶质与沥青质的1H NMR分析中以氘代氯仿为溶剂、四甲基硅烷为内标物。图11为空白试验油样和水热催化改质后油样的胶质和沥青质的1H NMR图谱。
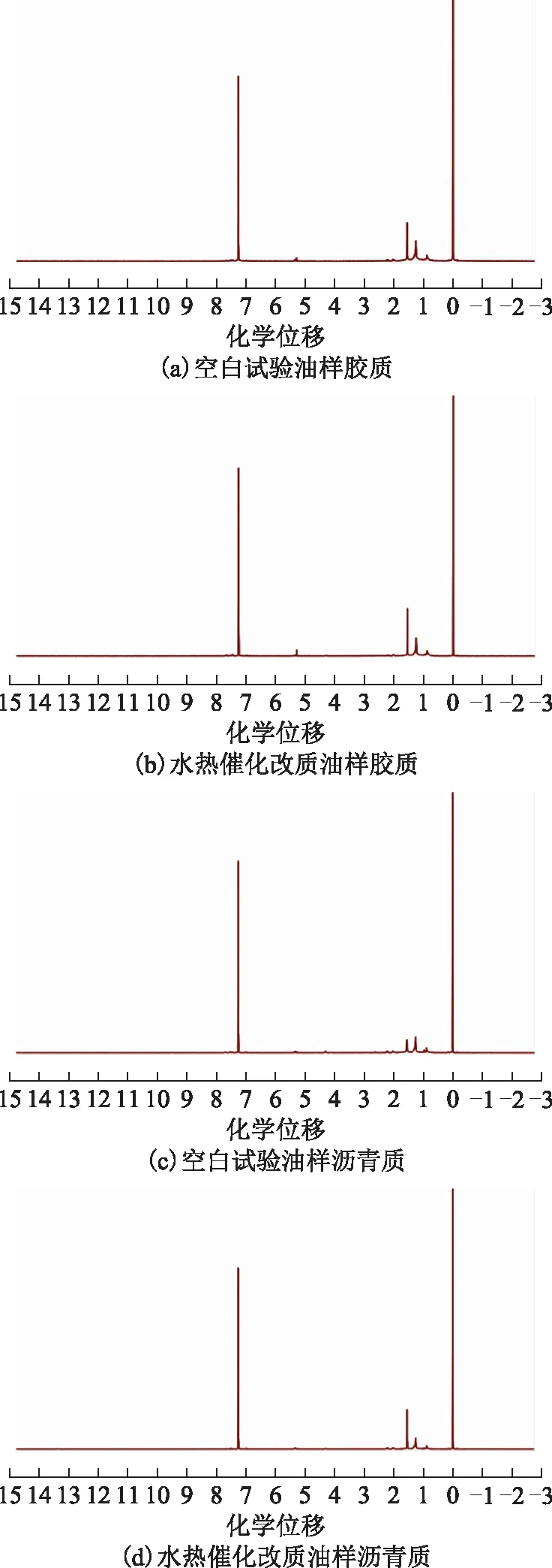
图11 空白试验油样和水热催化改质后油样的胶质和沥青质的1H NMR图谱
表5为根据图11中质子氢归属得到的结构参数[17],其中:HA为与芳香碳直接相连的氢原子数比例;Hα为与芳香环的α碳相连的氢原子数比例;Hβ为与芳香环的β碳上以及β以远的—CH2、—CH基上的氢原子数比例;Hγ为与芳香环的γ碳相连的—CH3基上的氢原子数比例。从表5可以看出,反应后胶质和沥青质的芳香度(fA)和芳香环系的缩合度参数(HAU/CA)降低。不饱和组分的加氢反应使得芳香性降低;芳香环系的缩合度参数的增加,可能是在水热催化裂解过程中发生了开环加氢反应,多环芳烃分子转化为重组分的小碎片所致[3]。并且,在反应后胶质、沥青质支化度(BI)指数均降低,表明在改质后胶质、沥青质结构中的支链数减少,这可能是大分子中部分侧链发生断裂,使得胶质、沥青质相对分子质量降低,从而降低了稠油黏度[18]。
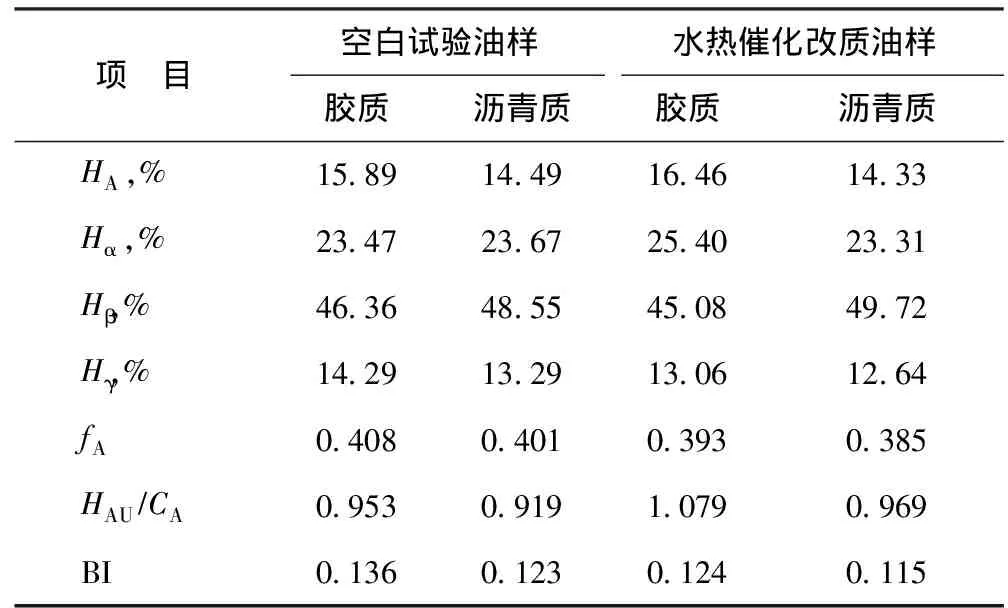
表5 空白试验油样与水热催化改质油样的胶质与沥青质的1H NMR结构参数
4 结 论
通过水热法成功合成出稠油水热催化改质降黏催化剂OA-Fe2(MoO4)3,并用于新疆克拉玛依稠油降黏试验,得出水热催化改质降黏试验的最佳工艺条件为:OA-Fe2(MoO4)3添加量0.4%,反应温度240 ℃,反应时间36 h,此时降黏率为61.21%。在反应过程中,重组分(胶质、沥青质)通过C—N,C—O,C—S键的断键,长链结构的断裂以及不饱和组分的加氢转变成轻组分(饱和分、芳香分),这可以从族组成、元素组成、fA及HAUCA得到验证,这解释了稠油黏度降低的原因,可为以后进一步的研究提供参考。
