不同产地赤芝药材的HPLC指纹图谱建立及聚类分析△
2019-07-03段晓颖陈霄马秋莹牛晓静翟海容
段晓颖,陈霄,马秋莹,牛晓静,翟海容
1.河南中医药大学第一附属医院,国家中医药管理局中药制剂三级实验室,河南 郑州 450000;2.河南中医药大学 药学院,河南 郑州 450000;3.商丘市中医院,河南 商丘 476000;4.河南中医药大学第一附属医院,国家中医药管理局中药制剂三级实验室,河南 郑州 450000;5.江苏正大天晴药业集团股份有限公司,江苏 南京 210000
中药现代化研究的重要任务之一就是建立一套客观的质量控制方法用以规范、指导中药材的炮制加工和中成药的生产。建立中药指纹图谱的目的就在于将中药质量控制模式与化学药品区别开来,充分体现中药的整体效应。中药指纹图谱是中药材经适当处理后,采用一定的分析手段和仪器测得的、能够标识该中药材各种组分群体特性的共有峰的图谱,是借用DNA指纹图谱发展而来的概念[1]。该技术是中药质量控制的有效解决方案,其中相似度的计算被认为是能够客观、全面地反映中药色谱指纹图谱之间相似情况的一种有效评价手段,计算相似度可以定量地衡量两指纹图谱之间的相似程度。本实验通过建立指纹图谱结合聚类分析法对10个产地的赤芝药材的不同指标变量进行分析,从而比较各个变量之间的相关性,将相似的变量提取出来,减少了分析变量的个数,使其更适用于各个变量间的相关分析[2-5]。
灵芝含有多种化学成分,如三萜类化合物、灵芝多糖、生物碱、核苷等有效成分,其中三萜类化合物为灵芝主要的有效成分,具有多种生理活性[6-7]。《中国人民共和国药典》2015版一部收载灵芝药材项下包括赤芝和紫芝两个品种,灵芝大部分为栽培品种且多为赤芝,该品种为多孔菌科真菌赤芝Ganodermalucidum(Leyss.ex Fr.)Karst.的干燥子实体。据报道,紫芝与赤芝的指纹图谱共有模式存在很大区别,在紫芝中未检测到灵芝酸A、灵芝酸B、灵芝酸C2、赤芝酸A、灵芝酸G、灵芝酸E 6种成分[8]。有报道称紫芝中含三萜酸的量明显比赤芝少[9],笔者在研究中也发现,赤芝中总三萜成分高于紫芝,因此确定赤芝这一品种作为研究对象。本文采用高效液相色谱法对不同产地赤芝药材进行分析,利用中药色谱指纹图谱相似度评价软件得到能够体现赤芝药材质量的指纹图谱,并进行聚类分析,为不同产地赤芝药材的鉴别和质量控制提供依据[10-12]。
1 材料
1.1 仪器
Aglient 1260高效液相色谱仪;AEL-200电子天平(日本岛津公司);Sartorius CP225D电子天平;HK250科导台式超声波清洗器(上海汉克科学仪器有限公司)。
1.2 试药
灵芝酸A(140324)、灵芝酸G(140504)(成都普菲德生物技术有限公司);乙腈、甲醇、磷酸为色谱纯;其他试剂均为分析纯。
赤芝药材经河南中医药大学第一附属医院陈天朝主任药师鉴定为多孔菌科真菌赤芝Ganodermalucidum(Leyss.ex Fr.)Karst.的干燥子实体。
2 方法与结果
2.1 10批赤芝药材来源及含量测定
按照2015版《中华人民共和国药典》一部灵芝项下方法测定10批不同来源赤芝中三萜及甾醇含量,见表1。

表1 10批赤芝药材批号、产地与三萜及甾醇含量 %
结果表明,三萜及甾醇含量符合《中华人民共和国药典》2015版规定(不得少于0.5%)。
2.2 色谱条件
色谱柱:ZORBAX SB-C18(250 mm×4.6 mm,5 μm),流动相:A乙腈-B 0.2%磷酸水,检测波长:254 nm;柱温:30 ℃;进样量:10 uL;流速:1.0 mL·min-1;信号采集时间:90 min,梯度洗脱程序:0~30 min,33%~34%A;30~60 min,34%~80%A;60~70 min,80%~100%A;70~75 min,100%~33%A;75~90 min,33%A[10]。
2.3 对照品溶液的制备
精密称取灵芝酸A、灵芝酸G对照品适量,加甲醇制成每1 mL含灵芝酸A 80.48 μg、灵芝酸G 86.40 μg的对照品溶液。
2.4 供试品溶液的制备
称取赤芝药材粉末约1 g,精密称定,精密加入无水乙醇50 mL,称定质量,回流60 min,放冷,称质量,用无水乙醇补足质量。滤过,取续滤液,用0.22 μm微孔滤膜滤过,备用[11]。
2.5 测定法
以灵芝酸A、灵芝酸G为参照物,照上述色谱条件测定并记录色谱图,利用“中药色谱指纹图谱相似性评价系统(2004A版)”软件构建10批样品色谱图,见图1~2。

注:1.灵芝酸G对照品;2.灵芝酸A对照品。图1 混合对照品HPLC图
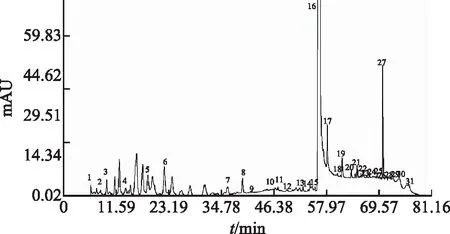
图2 赤芝HPLC指纹图谱
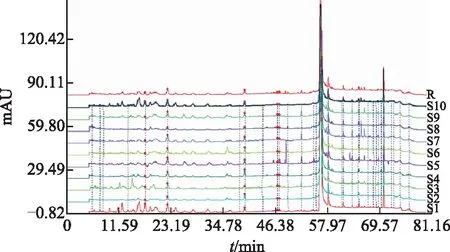
图3 10批赤芝药材HPLC图
2.6 方法学考察
2.6.1 精密度试验 取批号为140304-1赤芝药材制备的供试品溶液,连续进样5次,记录各共有色谱峰的保留时间和峰面积,各主要色谱峰相对保留时间和相对峰面积比值无明显变化,RSD分别为0.01%~0.28%和00.06%~2.74%,说明精密度良好[12]。
2.6.2 重复性试验 取批号为140304-1赤芝药材5份,分别制备成供试品溶液,进样测定,记录各共有峰的保留时间和峰面积,各主要色谱峰相对保留时间和相对峰面积比值无明显变化,RSD分别为0.01%~0.45%和0.14%~2.86%,结果表明重复性良好。
2.6.3 稳定性试验 取上述赤芝药材制备的供试品溶液,分别在0、6、12、18、24、30 h进样,记录各共有峰的保留时间和峰面积,结果表明,各主要色谱峰相对保留时间和相对峰面积比值无明显变化,RSD分别为0.01%~0.19%和0.35%~5.31%,结果表明稳定性良好。
2.7 不同产地赤芝药材指纹图谱的构建和相关技术参数
在上述条件下,共测定了10批赤芝药材,标定出31个共有峰,见图1~2。其中10批样品共有峰面积占总峰面积比值平均值为71.77%,非共有峰面积占28.23%,对比混合对照品的各色谱峰保留时间,确定2个主成分色谱峰[13]。在31个共有峰中,5号峰为灵芝酸G,6号峰为灵芝酸A。其中6号峰在10批样品中的峰面积和保留时间均较稳定,且分离较好,故选择6号峰为参照峰S,计算各色谱峰的相对保留时间和相对峰面积[14],见表2~3。

表2 10批赤芝药材指纹图谱共有峰相对保留时间

续表2

表3 10批赤芝药材指纹图谱共有峰相对峰面积

续表3
2.8 数据处理
2.8.1 不同产地赤芝药材相似度分析 利用“中药色谱指纹图谱相似度评价系统(2004A版)”软件,将实验数据导入,以相关系数(中位数和平均数)代表其相似度,10批赤芝药材的相似度结果见表4。
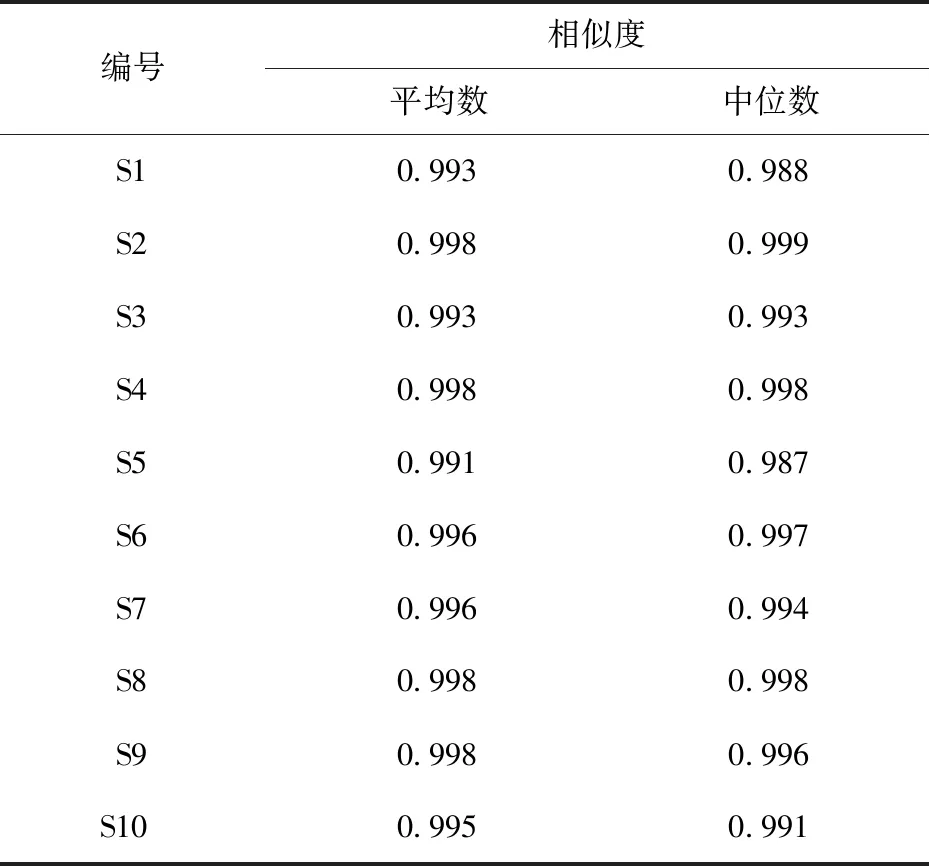
表4 10批赤芝药材指纹图谱相似度结果
由表3可知,不同方法所得相似度结果趋于一致,10批样品之间相似度均在0.9以上,符合中药色谱指纹图谱相似度技术要求。
2.8.2 不同产地赤芝药材聚类分析 对10批不同产地、不同批号的赤芝做指纹图谱分析,获得了包括灵芝酸G(5号峰)、灵芝酸A(6号峰)在内的31个共有色谱峰[15],将各共有色谱峰相对于参比峰的峰面积进行量化,得到10×31阶原始数据矩阵,代入SPASS20.0统计软件,采用系统聚类法,以欧式距离为测度[16],对10批赤芝药材各共有峰的峰面积进行分析,生成平均联接组间树状图,见图4,聚类分析将10批药材分成4类,其中3、4、5、6、7、9号样品聚为第一类,该类样品均为安徽产赤芝药材[17];1号和10号样品聚为第二类,2号样品单独聚为第三类,1、2、10号样品均为山东产赤芝药材,但聚类分析并未聚为一类,可能因为2号样品的16号、17号峰的相对峰面积与1、10号样品有一定差距;8号样品为湖南产赤芝药材,聚为第四类,从相对峰面积可以看出8号样品的5、16、17、19、27、30、31号峰均与其他批号的产品峰面积相差较大,因此将其单独聚为一类[18]。该聚类分析说明不同产地灵芝药材中所含三萜类成分相差较大,产地之间有显著性差异。

图4 10批灵芝药材聚类分析树状图
3 讨论
3.1 提取方式和提取溶剂的选择
本文首先考察了超声和回流两种提取方法,结果发现,回流提取检测出的色谱峰较多,故选择回流提取方法;选用三氯甲烷、甲醇、乙醇3种溶剂进行提取,发现三氯甲烷提取时,仅有少量峰被检测出来,甲醇回流提取时杂质峰较多,灵芝酸A未能和其他峰达到较好分离,而无水乙醇提取时灵芝酸A能得到较好分离,因此选择无水乙醇回流提取。
3.2 流动相的确定
本实验分别采用了5个流动相系统:乙腈-0.2%磷酸水;乙腈-0.03%磷酸水;乙腈-0.04%甲酸水;乙腈-0.1%甲酸水;乙腈-0.1%醋酸水。结果表明,乙腈-0.2%磷酸水系统为流动相,洗脱出色谱峰较多,且各色谱峰分离效果较好,因此选择乙腈-0.2%磷酸水系统为流动相。
3.3 柱温的选择
柱温的选择是影响指纹图谱的重要因素之一。本实验首先在20 ℃柱温下进行洗脱,结果发现,该条件下基线漂移非常严重,而选择30 ℃柱温进行洗脱,基线基本平稳,各色谱峰也能较好地被洗脱出来。
3.4 检测波长的选择
采用Agilent 1260 DAD检测器对样品进行全波长扫描,综合考虑参照峰(3、4)和其他成分的吸收,根据3D-plot图谱,在254 nm处检测的图谱信息量较多,峰形较好,mAU值较大,溶剂干扰少,基线平稳,因此选择检测波长为254 nm。
3.5 洗脱程序的优化
赤芝药材成分复杂,报道分离出的三萜类成分就有150余种,且含量较低,等度洗脱很难在保证较好分离度的情况下完全洗脱出来,故采用梯度进行洗脱分离。分离时,初始乙腈比例为25%时,各成分峰的Rt较大,前45 min内几乎无峰,扩大有机相比例为33%时,各成分峰的Rt大大缩小,对照品灵芝酸G在15 min左右出峰,灵芝酸A在22 min左右出峰,洗脱90 min将绝大部分三萜类成分洗脱出来,因此以初始有机相33%的比例进行洗脱,筛选出最佳梯度:0~30 min,33%~34%乙腈;30~60 min,34%~80%乙腈;60~70 min,80%~100%乙腈;70~75 min,100%~33%乙腈;75~90 min,33%乙腈。
4 总结
本文分别用中位数和平均数计算了10批赤芝样品的相似度,结果显示,3个产地10批样品之间相似度均较高,未将质量区别开,但通过聚类分析发现,赤芝质量与产地具有较高相关性,基本上同一产地可以聚为一类,不同产地之间质量还是有差别的。聚类分析与相似度分析结果并不完全相同,说明仅靠相似度分析并不能作为赤芝药材质量鉴别的精细与科学的方法。本文利用相似度结合聚类分析,为赤芝药材的质量评价提供了可靠依据,也为其质量控制提供了更全面的信息。检索多篇关于赤芝药材指纹图谱文献,发现将指纹图谱与聚类分析相结合对其产地进行分析和归属的报道较少,因此本文对赤芝不同产地的质量研究具有一定的指导意义。
