乙酰辅酶A羧化酶抑制剂的研究进展
2019-07-03梅连阔魏强强张惠斌周金培
梅连阔,魏强强,张惠斌*,周金培
(中国药科大学 1新药研究中心;2药物化学教研室,南京 210009)
随着人们生活水平的不断提高,非酒精性脂肪肝病(non-alcoholic fatty liver disease,NAFLD)在全世界的发病率不断上升,已经成为威胁人类健康的巨大隐患[1],在我国更成为仅次于病毒性肝炎的第二大肝病[2-3]。该病是指除酒精和其他明确的损肝因素所致的肝细胞内脂肪过度沉积为主要特征的临床病理综合征[4],包括单纯性脂肪肝(simple fatty liver,SFL)、非酒精性脂肪肝炎(non-alcoholic steatosis hepatitis,NASH)及其相关肝硬化[5]。1953年,Medes等[6]研究发现肿瘤细胞具有脂肪代谢异常旺盛的特点;且相比于正常细胞主要优先依靠外源摄取满足自身脂肪需求[7],肿瘤细胞的脂肪则主要来源是自身胞内的全新脂肪合成(de novo lipogenesis,DNL)[8],其合成速率与正常人体脂肪合成最旺盛的肝脏相当。脂肪酸参与肿瘤形成主要有3个方面:(1)参与肿瘤细胞膜磷脂的形成[9];(2)为肿瘤细胞增殖和存活提供能量;(3)用于合成一系列促肿瘤生长的脂质信号分子[10]。因此,寻找调控脂肪代谢的有效靶点,开发研究可以有效改善脂肪代谢异常的药物显得尤为重要。乙酰辅酶A羧化酶(ACC)作为脂肪酸合成代谢第一步反应的限速酶和关键酶,在肥胖、糖尿病、非酒精性脂肪肝病以及肿瘤的发生、发展等方面起着至关重要的作用[11-12]。ACC的抑制主要是通过抑制脂肪酸合成和促进脂肪酸的氧化从而发挥作用[13]。本文将对ACC的结构特点、作用机制及其抑制剂的研究进展进行综述。
1 ACC的结构和生物学功能
1.1 ACC的结构和功能
ACC存在于胞液中,是一种依赖生物素的变构羧化酶,主要催化依赖ATP的乙酰辅酶A(acetyl-CoA)转化为丙二酰辅酶A(malony-CoA)[14](图1)。其结构主要由羰基转移酶域(carboxyl transferase domain,CT)、生物素羧基载体蛋白(biotin carboxy carrier protein,BCCP)和生物素羧化酶域(biotin carboxylase domain,BC)组成[15]。乙酰辅酶A转化为丙二酰辅酶A的生理过程如下:首先,在ATP的参与下,碳酸氢根离子与结合在BC域的生物素生成生物素-羧酸盐复合物[16],后者经过一狭窄的蛋白酶体通道转运至CT域,羧酸根离子由生物素-羧基复合物转移至乙酰辅酶A,从而生成丙二酰辅酶A[17]。
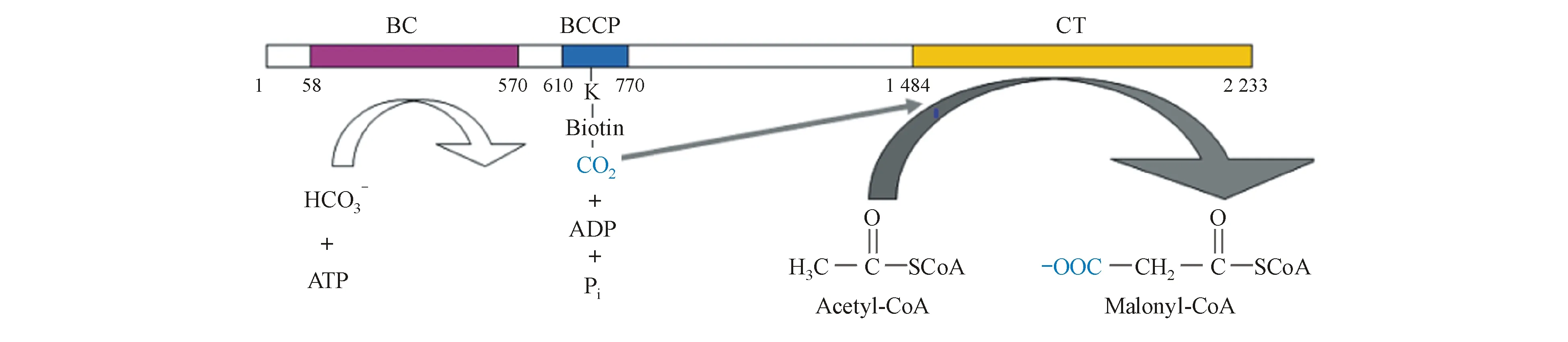
图1 ACC单体的结构组成及丙二酰单酰辅酶A的生成
BC:生物素羧化酶域;BCCP:生物素羧基载体蛋白;CT:羰基转移酶域;ATP:腺嘌呤核苷三磷酸;ADP:二磷酸腺苷;Acetyl-CoA:乙酰辅酶A;Malonyl-CoA:丙二酸单酰辅酶A
ACC有两种亚型:ACC1和ACC2,分别由不同的基因编码,且分布迥异[18]。ACC1存在于细胞质内,主要高表达于脂肪生成活跃组织,如肝脏、脂肪组织、乳腺等,调节脂肪酸的合成[19];其在这些组织内催化生成的丙二酰辅酶A可作为长链脂肪酸从头合成碳链延长的C2供体,进而合成三酰甘油和磷脂[20]。ACC2则主要存在于线粒体外膜上,高表达于脂肪氧化活跃组织,如骨骼肌、心脏等,调节脂肪酸的氧化[21];其在这些组织催化生成的丙二酰辅酶A是脂肪酸氧化的有效抑制剂,因为长链乙酰辅酶A通过肉毒碱棕榈酰转移酶1(carnitine palmitoyl transferase 1,CPT-1)转运至线粒体内,从而进行β氧化,而ACC2催化生成的产物丙二酰辅酶A是CPT-1的有效抑制剂[22],故而起到抑制脂肪酸氧化的作用。
1.2 ACC的抑制机制
细胞内ACC的调节机制较为简单,主要受上游腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)磷酸化调控[23]。ACC二聚化是其活性所必需的,BC结构域的C末端有一段氨基酸侧链,上游的AMPK可以磷酸化该氨基酸侧链上的Ser117,使得该侧链折叠并与Arg277结合,阻碍ACC二聚体的形成,从而抑制ACC的活性[24]。
由前所述,丙二酰辅酶A的生成需由BC域和CT域共同参与;故而对ACC的抑制可以通过化学小分子对BC或者CT结构域的反应来实现。先以化合物ND-630为例,其作为BC结构域的抑制剂,既不会直接激活AMPK活性,亦不会阻碍AMPK对ACC的磷酸化,而是直接与Arg277形成关键氢键作用,模拟磷酸化后的Ser222,阻碍ACC二聚化的发生。其次以化合物CP-640186为例,其作为CT结构域的抑制剂,通过占据生物素-羧基复合物在乙酰基转化为丙二酰基过程中作用于CT域的位置,从而阻断羧化反应的发生而起到抑制ACC活性的作用[25]。
2 ACC抑制剂的研究进展
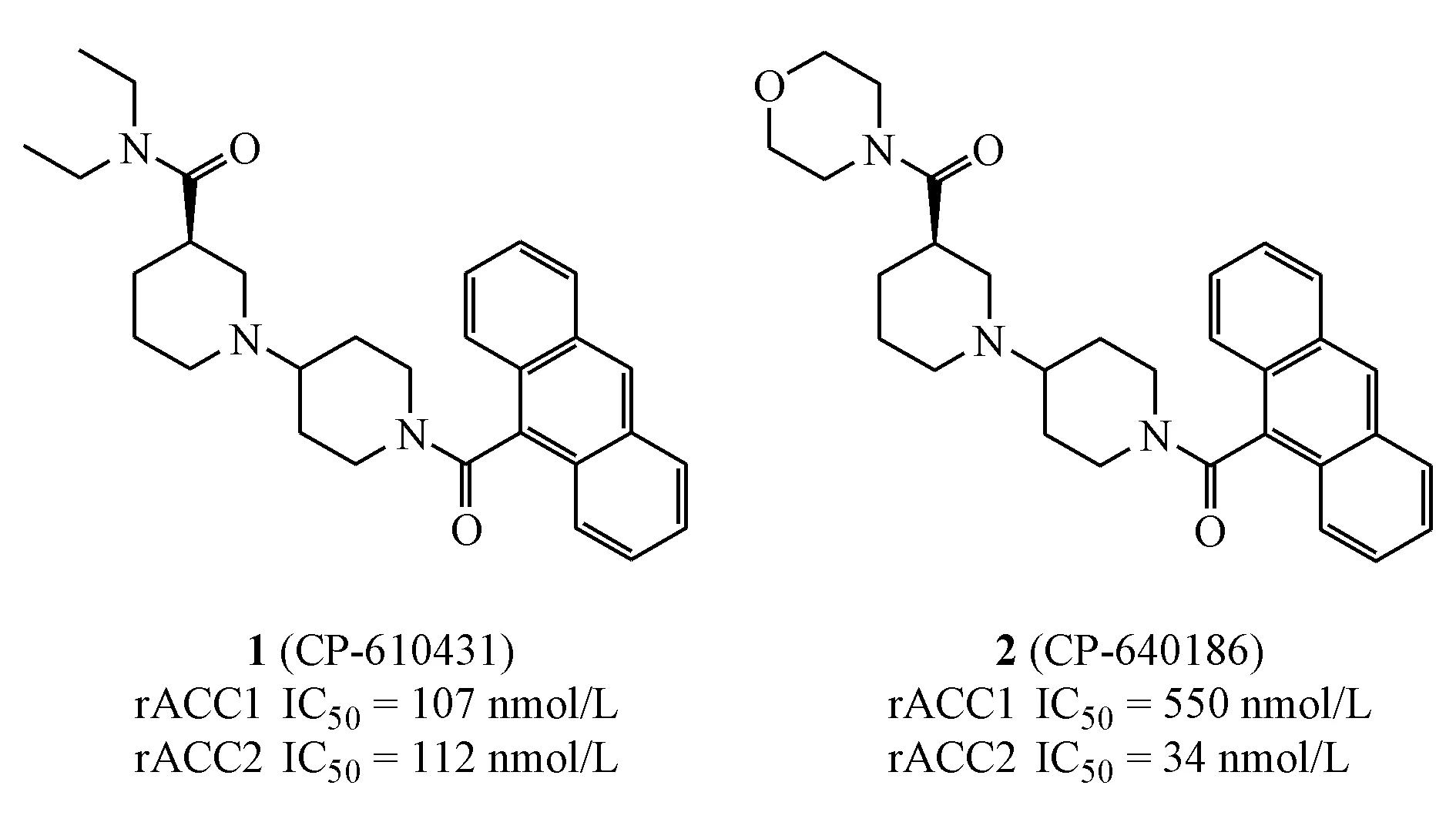
迄今为止,虽无小分子ACC抑制剂作为治疗药物上市,但ACC在脂肪代谢方面的突出作用,使其成为寻找新型治疗非酒精性脂肪肝病等代谢性疾病和肿瘤的热门靶点。自2003年Pfizer公司公布第1个小分子ACC抑制剂CP-610431(1)以来,时至今日各大制药公司如Gliead、Nimbus、Bayer、Takeda、Boehringer Ingelheim等对该靶点的临床研究正努力进行中,现有处于临床Ⅱ期研究2个、临床Ⅰ期研究1个、临床前研究1个,且都处于活跃研究状态,有多个小分子处于生物活性评价阶段。治疗的疾病多涉及肝脏代谢性疾病(NASH等)、肥胖、2型糖尿病(T2DM)以及非小细胞肺癌(non-small cell lung cancer,NSCLC)等。现根据文献报道的ACC抑制剂按其在研公司分类并进行综述。
2.1 Pfizer公司
2003年,Pfizer公司通过计算机高通量筛选得到第1个小分子ACC抑制剂CP-610431(1)(大鼠肝脏ACC1 IC50=107 nmol/L,大鼠骨骼肌ACC2 IC50=112 nmol/L),并以此为结构基础进行结构优化得到2(CP-640186)(大鼠肝脏ACC1 IC50=550 nmol/L,大鼠骨骼肌ACC2 IC50=34 nmol/L)。化合物CP-640186不仅可以促进C2C12细胞内脂肪酸氧化(ACC2 EC50=57 nmol/L),明显降低小鼠体内肝脏、比目鱼肌、四头肌、心肌的丙二酰辅酶A水平(EC50分别为55,6,15,8 mg/kg),还可以抑制正常小鼠、CD1小鼠、ob/ob小鼠体内脂肪酸的合成(EC50分别为13,11,4 mg/kg)[26]。后续研究进一步揭示了CP-640186与ACC的结合位点及方式[27],共晶(PDB:1W2X)显示该化合物嵌入到ACC二聚体的夹缝中,并与Glu-2026和Gly-1958形成关键氢键作用,通过阻断生物素底物与CT域的结合而抑制ACC的活性。
为进一步降低化合物的熵以提高分子与蛋白的亲和力,Pfizer公司参照CP-640186与ACC的结合方式,开发了螺苯并二氢吡喃酮类ACC抑制剂。化合物3在表现出较好的ACC抑制活性的同时,也具有较高的肝微粒体清除率(human liver microsomal clearance,HLM)(hACC1 IC50=22 nmol/L,hACC2 IC50=48 nmol/L,lgD (pH=7.4,下同)=3.8;CLint=86 mL·min-1·kg-1)。故以芳杂环取代亲脂性较高的苯环得到化合物4(rACC1 IC50=17 nmol/L,hACC2 IC50=11 nmol/L;lgD=1.8;CLint<8 mL·min-1·kg-1)和化合物5(PF-1027)(hACC1 IC50=7.0 nmol/L,hACC2 IC50=6.9 nmol/L),在提高抑制活性的同时,很大程度上降低了该化合物的固有清除率。将化合物4中的二氢吡喃酮部分替代为螺内酰胺,并对右侧羧酸取代基进行优化,得到化合物6(hACC1 IC50=10 nmol/L,hACC2 IC50=4 nmol/L;iv 1 mg/kg CLint=1.7 mL·h-1·kg-1,F=71%)。该化合物与ACC的结合方式及位点与CP-640186完全相同。以化合物4为例,通过共晶(PDB:4WYO)可以发现化合物与酶的作用位点位于ACC二聚体的夹缝中间,并与氨基酸Gly1958和Glu2026存在关键氢键作用力(图2[29])。
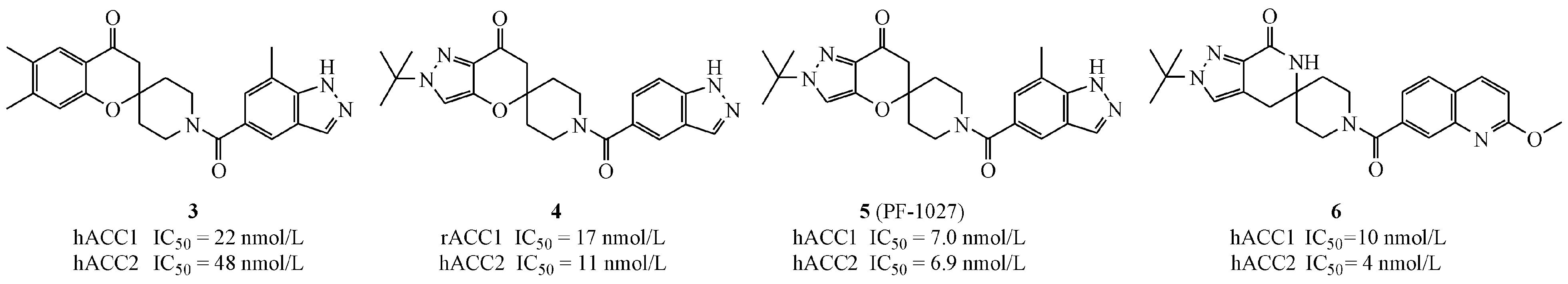
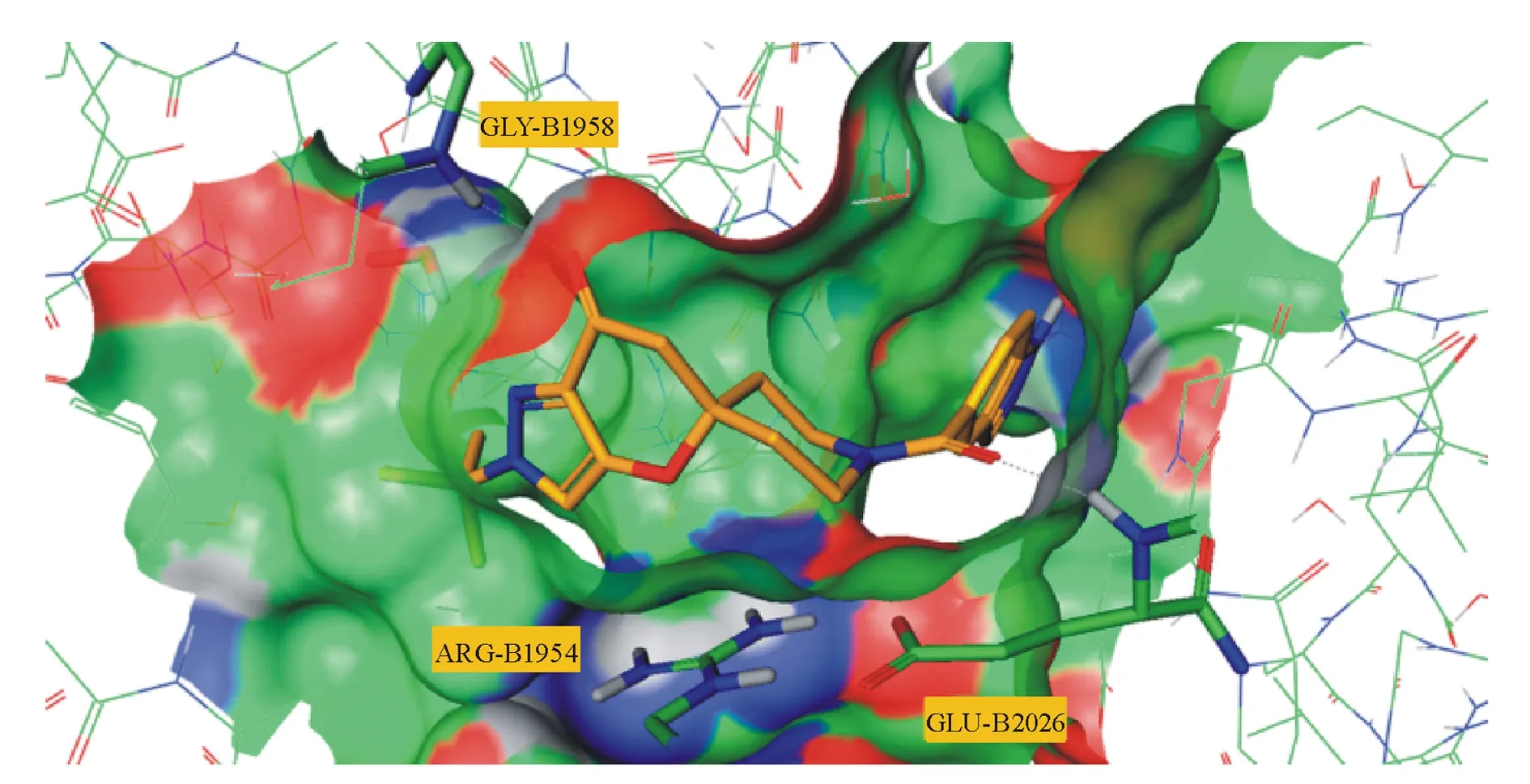
图2化合物4与ACC CT域的结合共晶结构
此后,在2010-2015年期间,Pfizer公司对螺苯并二氢吡喃酮类ACC抑制剂的二氢吡喃酮部分进行了结构拓展,并进行了多篇专利保护,代表化合物7~10,酶抑制活性的IC50均小于100 nmol/L。其中化合物7(PF-05175157)(hACC1 IC50=98 nmol/L,hACC2 IC50=45 nmol/L)于2015年1月被推上临床,现处于临床Ⅰ期研究中,主要用于2型糖尿病的治疗研究[28]。此外,2017年11月,Pfizer公司公开了化合物11(PF-06423264)(螺环类似物,结构未知)的临床Ⅰ期和化合物12(PF-05221304)(螺环类似物,结构未知)的临床Ⅱ期试验进程,分别用于痤疮和肝脏、胆管相关疾病的治疗研究;且化合物12被FDA给予绿色通道,以加速其用于NASH的临床研究进程,但化合物11因用于治疗痤疮失败,已被从临床试验撤回。Pfizer公司尚未公开这两个化合物的任何药理或临床数据。值得注意的是,该类化合物的临床试验表明,受试者血浆中三酰甘油的水平明显高于对照组,其机制尚不明确,可能与ACC CT域的结合抑制有关[29]。
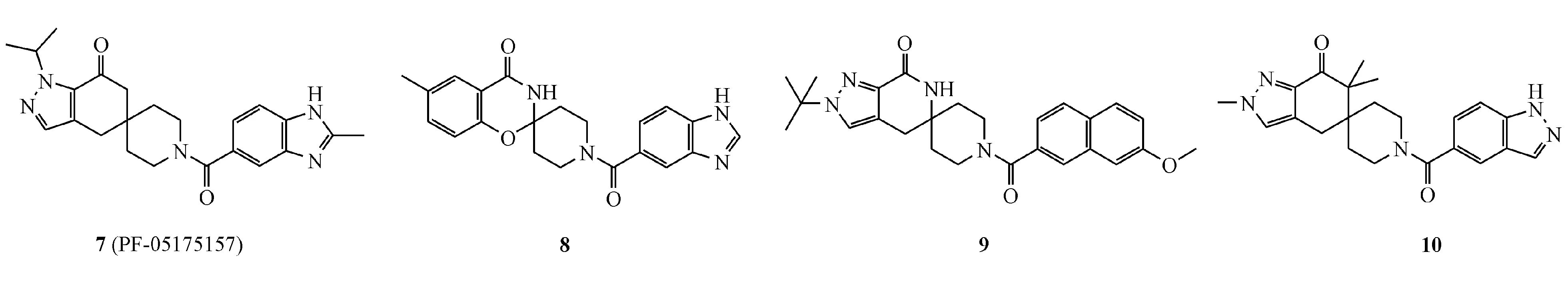
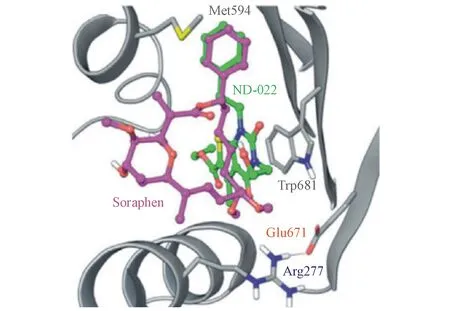
图3 ND-022、Soraphen A与hACC2 BC域结合的共晶结构(ND-022:绿;Soraphen A :粉)
2.2 Nimbus Therapeutics公司
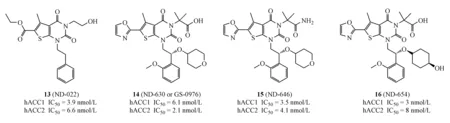
2013年,Nimbus公司参照Soraphen A与ACC的结合位点以及作用方式,通过计算机高通量筛选得到先导化合物13(ND-022)(hACC1 IC50=3.9 μmol/L,hACC2 IC50=6.6 μmol/L);共晶显示(图3[30]),ND-022与Soraphen A都结合在ACC BC域,结合位点重叠。研发人员在化合物13结构的基础上,进行药物设计,优化分子与蛋白之间非共价结合作用力,运用生物电子等排进行元素替换以期获得良好的类药性质,从而得到化合物14(ND-630,NDI-010976,GS-0976,hACC1 IC50=6.1 nmol/L;hACC2 IC50=2.1 nmol/L);这是第1个结合于BC域、且表现出优异活性和良好类药性质的小分子ACC抑制剂[30]。该化合物在HepG2细胞水平能有效抑制脂肪酸从头合成(IC50=66 nmol/L);正常大鼠灌胃给药实验中,可明显降低丙二酰辅酶A的含量(ED50=1.5 mg/kg);在SD大鼠灌胃给药实验中,可明显降低呼吸熵(respiratory quotient,RQ)和脂肪酸的合成(ED50=0.14 mg/kg);在由高脂饮食诱导的肥胖大鼠为期28 d灌胃给药实验中,可明显降低胰岛素含量、提高胰岛素敏感性、降低肝脏和血液中三酰甘油和胆固醇的水平,且并未发现明显的丙氨酸转氨酶水平的改变或肝质量减轻[31]。化合物14最先由Nimbus公司于2014年推向临床,用于NASH的治疗。随后2016年,鉴于治疗NASH的药物缺乏以及临床Ⅰ期疗效显著,美国FDA给予绿色通道,以期加速审批和临床试验。
与此同时,Nimbus公司又推进了化合物15(ND-646,hACC1 IC50=3.5 nmol/L;hACC2 IC50=4.1 nmol/L)和16(ND-654,hACC1 IC50=3 nmol/L;hACC2 IC50=8 nmol/L)分别在肿瘤领域,尤其是非小细胞肺癌(NSCLC)和肝癌的临床研究中。研究证明,化合物15可以明显降低脂肪酸合成从而抑制NSCLC细胞的生长迁移和异种移植或通过基因突变致癌小鼠体内肿瘤的生长,且在为期4周的100 mg/kg大剂量灌胃给药情况下并未表现出明显的不良反应[32];该化合物同样由Nimbus公司于2017年推向临床研究,主要用于非小细胞肺癌的治疗。化合物16主要用于原发性肝癌(hepato-cellular carcinoma,HCC)的实验研究,研究证明化合物16是一种新型、肝脏特异性的ACC抑制剂,其模拟ACC磷酸化的效应,抑制从头脂肪酸合成(DNL)和HCC的发展;同时证明了与激酶抑制剂索拉非尼联用能有效降低HCC的发生,并提高大鼠的存活率。这些数据表明,化合物16可能对治疗HCC患者有价值,特别是对HCC的S2亚型患者[33]。
2013-2017年期间,Nimbus公司先后公开了10篇专利,对该类化合物的母核结构进行了广泛拓展与保护,其所涉及母核结构种类多达128个。蛋白共晶(5KKN)显示,化合物ND-646同样结合于ACC的BC域,与AMPK磷酸化ACC的作用位点完全相同,且与Soraphen A(PDB:1W96)类似。
2.3 Gliead公司
2016年4月,Gliead公司以12亿美元的交易金额收购了Nimbus Therapeutics全资子公司Nimbus Apollo及其旗下在研乙酰辅酶A羧化酶抑制剂的项目,其中就包括化合物14(ND-630,GS-0976),这势必进一步拓展了该公司在非酒精性脂肪肝炎、原发性肝癌等在内的肝脏疾病研发管线[34]。在一随机双盲试验(临床Ⅰ期)中,通过持续10 h给30名(分为3组,每组10人)轻微肥胖的健康受试者口服灌注果糖(剂量:每30分钟150 mg/kg),使受试者体内脂肪合成速率高出正常水平30.9%,之后分别按照剂量20,50,200 mg/kg口服给药后,受试者体内脂肪合成分别减少70%、85%、104%,表现出明显的剂量依懒性,且在200 mg/kg高剂量口服给药时,受试者亦表现出良好的耐受性[35]。
为了评估化合物14作为ACC抑制剂在肝脏中的安全性和有效性,2018年7月27日,研究人员公布了一项针对NASH患者的临床Ⅱ期随机安慰剂对照实验[36],分析了来自126例肝脂肪变性至少8%的患者数据。从2016年8月8日到2017年7月18日,患者被随机分配到GS-0976 20 mg组,GS-0976 5 mg组和安慰剂组,持续给药12周。研究人员评估肝脂肪变性、硬度、血清纤维化标志物和血浆代谢组学的标志物的测量值,结果显示,给予化合物1420 mg组中48%的患者、GS-0976 5 mg 组中23%的患者和安慰剂组中15%的患者磁共振成像评估质子密度脂肪分数(MRI-PDFF,PDFF 反应)至少相对减少30%;磁共振弹性成像测量的硬度变化在各组之间没有差异,但将化合物14以20 mg/kg的剂量给予患者时,发现纤维化标志物(金属蛋白酶1)呈现剂量依赖性降低的现象。在将GS-0976以20 mg/kg的剂量给予患者时,酰基肉碱的血浆浓度也降低。这些数据证明,化合物14是安全的,但在给予化合物14的患者中观察到血清三酰甘油水平的平均值相对增加11%和13%。研发人员需要更深入地评估化合物14在NASH患者中的安全性和有效性。
2.4 Bayer公司
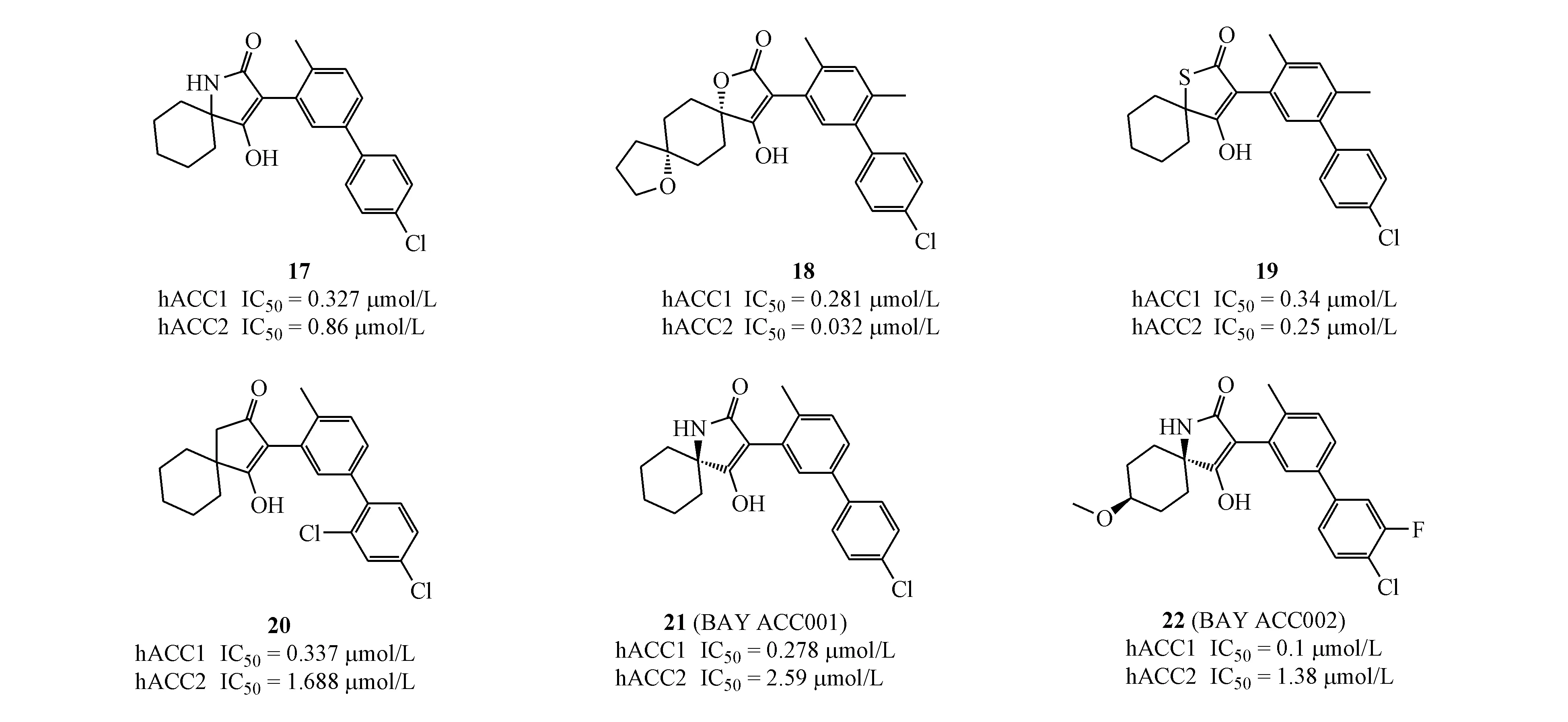
早在2002年,Bayer公司就曾公开过两篇ACC抑制剂的专利,其用途主要用作杀虫剂或者除草剂。鉴于近些年ACC抑制剂在代谢性疾病以及肿瘤方面的突出作用和快速研发进展,2012-2014年期间,Bayer公司连续公开4项专利,进一步保护与之前专利具有类似结构类型的ACC抑制剂,其用途涉及脂肪肝、肥胖、糖尿病、高脂血症以及肿瘤等,代表化合物为17~22。2016年,Bayer公司在Oncotarget杂志上发表了一篇ACC抑制剂的研究性文章,其中,化合物BAY ACC001(21)(hACC1 IC50=0.278 μmol/L,hACC2 IC50=2.59 μmol/L;在MCF-7细胞系中,抑制丙二酸单酰辅酶A生成的IC50为62 nmol/L)和BAY ACC002(22)(hACC1 IC50=0.1 μmol/L,hACC2 IC50=1.38 μmol/L;在MCF-7细胞系中,抑制丙二酸单酰辅酶A生成的IC50为32 nmol/L)主要通过抑制活性配体的棕榈化,从而发挥调控WNT和Hedgehog信号通路的作用,后者在诸多肿瘤如胰腺癌、宫颈癌以及肺癌中有异常表达[37]。该类化合物并未明确作用于ACC的位点或结合方式,且未见进一步研发报道。
2.5 Takeda公司
Takeda公司有关ACC抑制剂的研发主要集中在两类结构。首先,Takeda公司以化合物2(CP-640186)为先导化合物,参照其与ACC的CT域的结合方式,设计两条改造思路:(1)适当调整吗啉酰胺键的伸展方向,以期加强酰胺羰基与Gly2162的氢键作用力;(2)在保持分子与Glu2230氢键作用的前提下,替换蒽环、增加氢键供体,以期可能形成新的氢键作用力以增强分子与蛋白的亲和力[38]。研发人员设计合成了具有螺内酯母核的ACC抑制剂,如化合物23(hACC1 IC50=32 nmol/L,hACC2 IC50=43 nmol/L;CLint=230 mL·min-1·mg-1;lgD=3.01),抑制活性较先导化合物CP-640186有了很大的提高。后续代谢试验中发现螺内酯类化合物具有较高的亲脂性和和肝微粒体清除率,故将螺内酯代替为螺内酰胺得到化合物24(hACC1 IC50=120 nmol/L,hACC2 IC50=18 nmol/L;CLint=66 mL·min-1·mg-1;lgD=2.61),将苯并噻吩部分代替为吡啶并噻吩得到化合物25(hACC1 IC50=78 nmol/L,hACC2 IC50=14 nmol/L;CLint=79 mL·min-1·mg-1;lgD=2.49),在保持ACC抑制活性的同时,亲脂性和肝微粒体清除率皆有相当程度的改善[39]。
随后,Takeda公司又相继开发了螺吡唑烷二酮类ACC抑制剂,如化合物26(hACC1 IC50=120 nmol/L,hACC2 IC50=20 nmol/L;CLint=12 mL·min-1·mg-1;lgD=2.03)。值得注意的是,研发人员在共晶结构中发现化合物苯并噻吩环的7′位附近有一个由Glu2236、Lys1967、Ala1964等氨基酸组成的狭小口袋,于是在7′位引入甲氧基得到化合物27(hACC1 IC50=21 nmol/L,hACC2 IC50=4.9 nmol/L;CLint=35 mL·min-1·mg-1;lgD=2.33);该化合物不仅活性有了相当大的提高,且在SD大鼠灌胃或注射给药实验中,表现出良好的药代动力学性质;在Wistar肥胖大鼠灌胃给药实验中,可剂量依赖性地增加脂肪酸氧化和抑制脂肪酸合成[40]。该类化合物自2007-2009年期间公开了4项专利,至今未见后续报道。
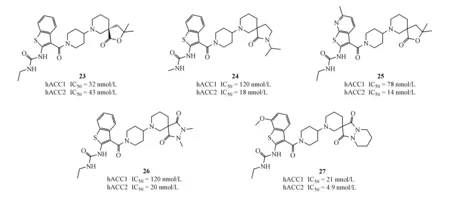
此外,在2010-2013年期间,Takeda公司公开了3篇专利,用以保护具有单环或二环结构特征的ACC抑制剂。随后于2015-2016年Takeda公司进一步拓展了该类化合物的专利保护范围。该类化合物的特点在于选择性抑制ACC1,而ACC1在肿瘤细胞中的表达远高于ACC2[32],Takeda公司也着重对该类化合物进行了抑制肿瘤活性方面的构效关系研究。研发人员以具有2-氮杂环丁基-1,3-苯并唑结构特征的化合物28(在浓度为10 μmol/L 时,对hACC1的抑制率为48%,对hACC2的抑制率为22%)为先导化合物,分别在左侧苯环及其取代基、中间连接链、右侧母核并杂环部分进行了构效关系研究,依次得到的代表性化合物为29(hACC1 IC50=220 nmol/L,hACC2 IC50>10 mmol/L)、30(hACC1 IC50=23 nmol/L,hACC2 IC50=5 800 nmol/L)、31(hACC1 IC50=230 nmol/L,hACC2 IC50>10 000 nmol/L)、32(hACC1 IC50=5.3 nmol/L,hACC2 IC50>10 000 nmol/L);其中活性最好的是化合物32(hACC1 IC50=5.3 nmol/L,hACC2 IC50>10 000 nmol/L;在pH为6.8的条件下,其溶解度小于0.21 μg/mL;对细胞色素P450酶系两个亚型CYP2C8和CYP2C9的抑制率分别为82.7%和89.9%)。
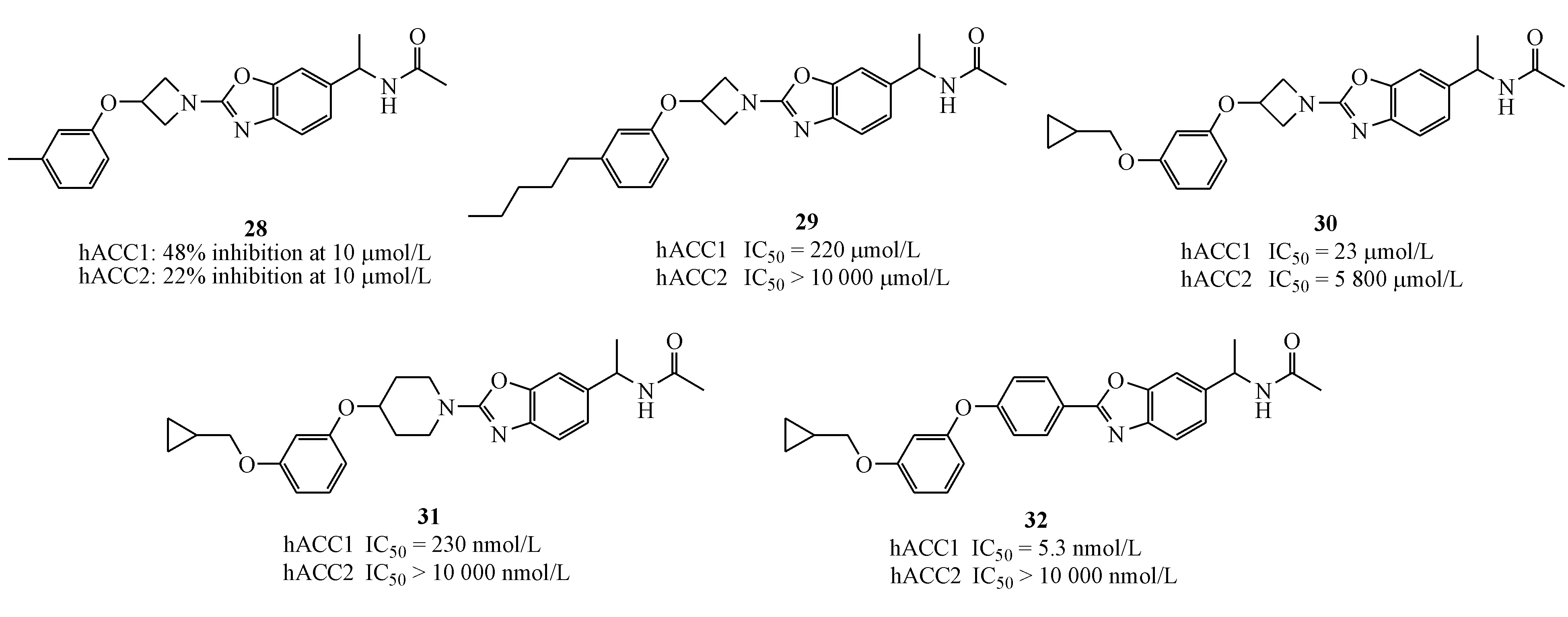
之后,在化合物32的基础上,研发人员希望在保持活性和选择性的同时,通过减少化合物中芳香环数量来改善其溶解性和降低对肝药酶(cytochrome P450,CPY)(10 μmol/L)的抑制活性,得到化合物33和34。然后以二者中较优的化合物34为基础,用醚链替代34中的烷基连接链,得到化合物35,为进一步改善单环唑类化合物的溶解性,将化合物35中间部分的苯环替换为吡啶环,将右侧乙酰胺部分替换为脲基,得到化合物36,极大的增强了对ACC和细胞的抑制活性,并取得了极好的选择性。后续研究表明,该化合物具有较低的肝微粒体清除率(hCLint=47 μL·min-1·mg-1;rCLint=16 μL·min-1·mg-1)和良好的代谢稳定性,并未发现明显的CPY抑制活性,且在正常小鼠给药实验(0.1 mg/kg,iv;1 mg/kg,po)都有优异的生物利用度(F=82.9%);在HCT116异种移植小鼠模型灌胃给药(单次,30 mg/kg,16 h)实验中,化合物36可持续有效地抑制丙二酰辅酶A的合成[41]。目前,该类化合物大多处于生物活性测试阶段,暂时未见更新报道。
2.6 Taisho公司
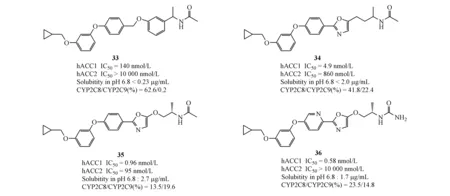
2009年,Taisho公司报道了一类ACC双重抑制剂[42],该类化合物同样是以化合物2(CP-640186)为先导化合物,将蒽环替换为2,6-二芳基吡啶,如化合物37(hACC1 IC50=101 nmol/L,hACC2 IC50=23 nmol/L),将2,6-二芳基吡啶替换为苯并噻吩脲得到化合物38。将化合物38中的哌啶上的乙酰基替换为Boc,得到化合物39(hACC1 IC50=24 nmol/L,hACC2 IC50=79 nmol/L),抑制活性有所增强,且在HepG2细胞水平可有效的抑制脂肪酸的合成。为了提高Boc基团的稳定性,进一步结构优化得到化合物40(hACC1 IC50=192 nmol/L,hACC2 IC50=95 nmol/L),且该化合物在HepG2细胞水平能有效促进脂肪酸氧化(EC50=370 nmol/L)、抑制脂肪酸合成(IC50=60 nmol/L);通过为期12 d高糖饮食诱导的SD大鼠每天两次剂量(3~10 mg/kg)灌胃给药实验显示,化合物40可明显降低肝脏以及血浆中三酰甘油的含量[43]。该类化合物至今未见更新报道。
2.7 Sanofi-Aventis/Amgen/Boehringer Ingelheim/Shionogi公司
鉴于这4家公司报道的ACC抑制剂结构类型相近,且相关专利和文献也较少,至今未见更新研究进展,故而一并列述。
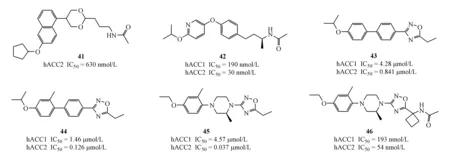
2010年,Sanofi-Aventis公司首先经过高通量筛选,得到化合物41(hACC2 IC50=630 nmol/L),该化合物只表现出微弱的ACC2抑制活性。进一步结构优化得到42(hACC1 IC50=190 nmol/L,hACC2 IC50=30 nmol/L;rACC1 IC50=170 nmol/L,rACC2 IC50=400 nmol/L);该化合物在Wistar大鼠体内表现出良好的药代动力学性质;此外,Wistar大鼠被灌注三酰甘油的同时,按50 mg/kg剂量灌胃给药可以有效促进大鼠体内脂肪酸氧化;通过给Zucker Diabetic Fatty(ZDF)大鼠每天按剂量30 mg/kg 灌胃给药实验证实,该化合物也可以有效降低三酰甘油水平[44]。
2013年,Amgen公司经过高通量筛选得到先导化合物43(hACC1 IC50=4.28 μmol/L,hACC2 IC50=0.841 μmol/L),进一步结构优化得到化合物44(hACC1 IC50=1.46 μmol/L,hACC2 IC50=0.126 μmol/L),为降低化合物44的亲脂性和改善类药性质[45],以哌嗪环替换中间苯环,得到45(hACC1 IC50=4.58 μmol/L,hACC2 IC50=0.037 μmol/L),虽然抑制活性有所提高,但该化合物在SD大鼠给药实验中代谢性质较差、生物利用度较低[iv (2 mg/kg)CLint=2.6 L·h-1·kg-1;po(5 mg/kg)cmax=380 nmol/L,AUC=291 ng·h/mL,F=17%],故而研发人员集中对结构中易代谢位点进行优化,得到化合物46(hACC1 IC50=193 nmol/L,hACC2 IC50=54 nmol/L;mACC1 IC50=427 nmol/L,mACC2 IC50=89 nmol/L),该化合物不仅体外及小鼠体内酶活优异,且在SD大鼠[iv (2 mg/kg)CLint=0.68 L·h-1·kg-1;po(5 mg/kg)cmax=1 200 nmol/L,AUC=2485 ng·h/mL,F=34%]和C67BL6小鼠[iv(2 mg/kg)CLint=0.21 L·h-1·kg-1;po(5 mg/kg)cmax=5 800 nmol/L,AUC=8 880 ng·h/mL,F=46%]给药实验中均表现出良好的药物代谢性质[46]。
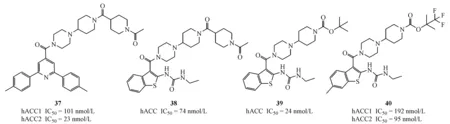
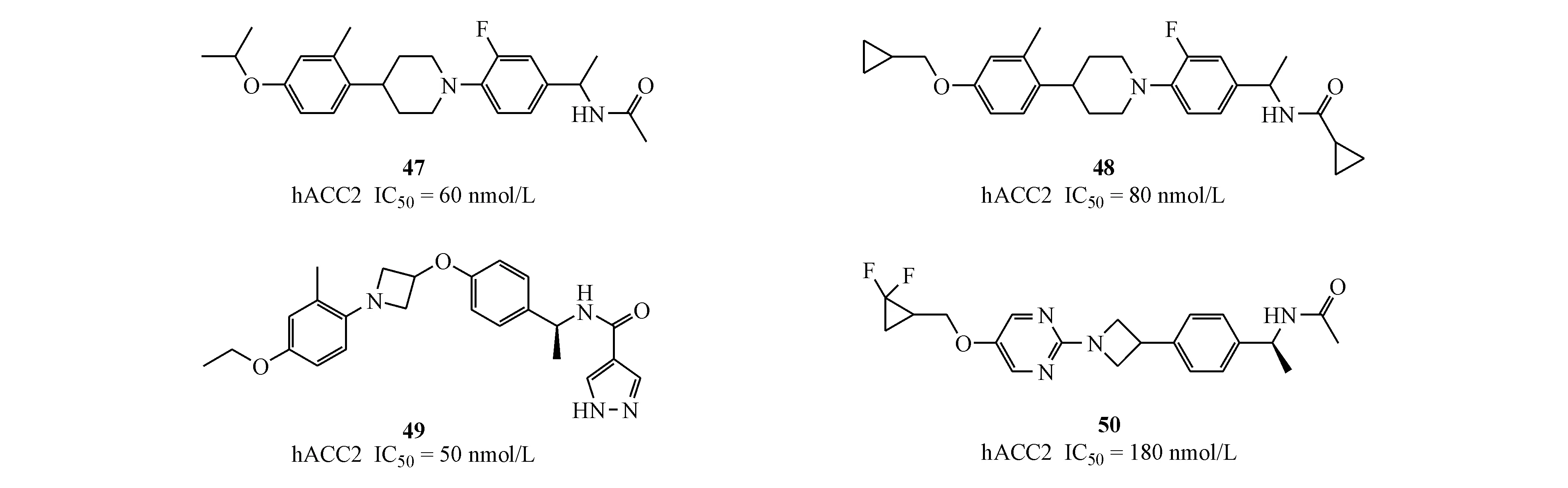
Boehringer Ingelheim 公司于2013年前后公开了4篇ACC抑制剂的相关专利。专利只报道了化合物对ACC2的抑制活性,代表化合物47~50。
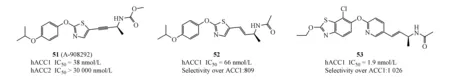
Shionogi公司在2018年发现新的烯烃衍生物,作为具有体内功效的选择性乙酰CoA羧化酶2抑制剂[47]。Shionogi公司基于Abbott公司报道的专一性的ACC2抑制剂51(A-908292)(hACC2 IC50=38 nmol/L,hACC1 IC50>30 000 nmol/L)结构特点,将炔烃替换为烯烃,得到化合物52(hACC2 IC50=66 nmol/L,其选择性高于hACC1 809倍;CYP2C9 IC50=4.4 μmol/L)成功规避了化合物51在麻醉大鼠中表现的严重癫痫和心血管危险因素,但该化合物抑制活性和选择性都明显下降。进一步结构改造修饰得到化合物53(hACC2 IC50=1.9 nmol/L,其选择性高于hACC1 1 026倍;CYP2C9 IC50=13 μmol/L),该化合物53在C57BL/6鼠体内PK实验中显示出低的血浆清除率和较高的生物利用度(0.2 mg/kg,iv,2.5 mg/kgpo;CLtot=0.354 mL·min-1·kg-1,F=83.5%),并且明显减少在该种鼠的骨骼肌中丙二酰辅酶A的水平(与对照相比,分别在1.0和2.5 mg/kg下有20%和41%丙二酰辅酶A生成减少)。目前,该化合物进一步的相关研究仍在进行中。
2.8 AstraZeneca公司
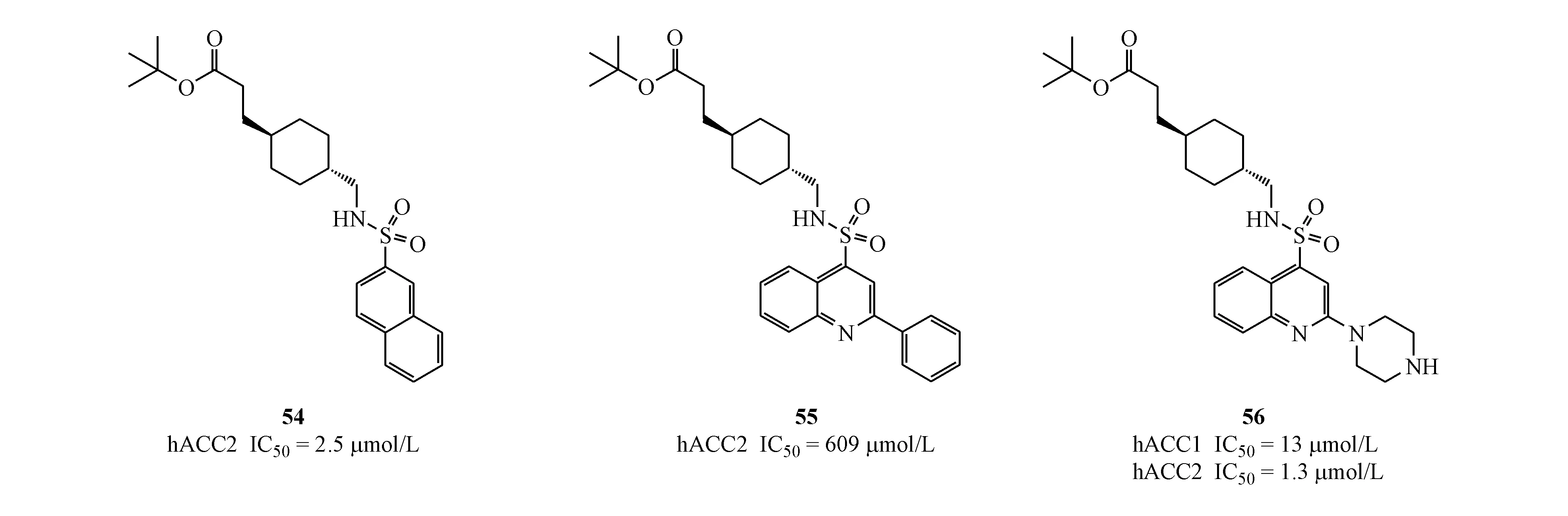
2010年,AstraZeneca公司研发团队以化合物2(CP-640186)为先导化合物设计了两条改造思路:(1)以其他芳环,如萘环、2-苯基喹啉、2-哌嗪喹啉等代替蒽环;(2)以环己基甲基芳磺酰胺替换化合物2中的4-哌啶基哌啶,试图在保留相似的空间构象的同时,维持与Gly2162和Glu2230的关键氢键作用,代表化合物如54~56。该类化合物虽在结构上有所突破,但ACC抑制活性有所下降,可能原因是脂肪链状Boc未能有效的与Glu2230形成稳固的氢键作用。且后续关于此类化合物的共晶显示,类似化合物2结构中4-哌啶基哌啶的特定空间构象对于活性的保持不可或缺,之后Pfizer研发的螺苯并二氢吡喃酮类ACC抑制剂也证实了这一观点[21]。该类化合物至今未见更新报道。
2.9 其他类
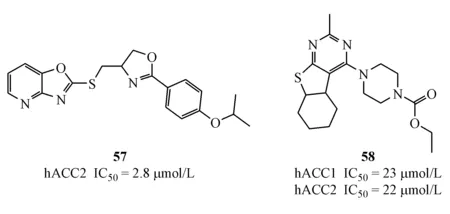
除上述ACC小分子抑制剂外,Haselkorn等[48]曾于2010年筛选得到两个化合物57(hACC1 IC50not determined;hACC2 IC50=2.8 μmol/L)和化合物58(hACC1 IC50=23 μmol/L,hACC2 IC50=22 μmol/L),只表现出微弱的ACC抑制活性,且之后再无更新报道。
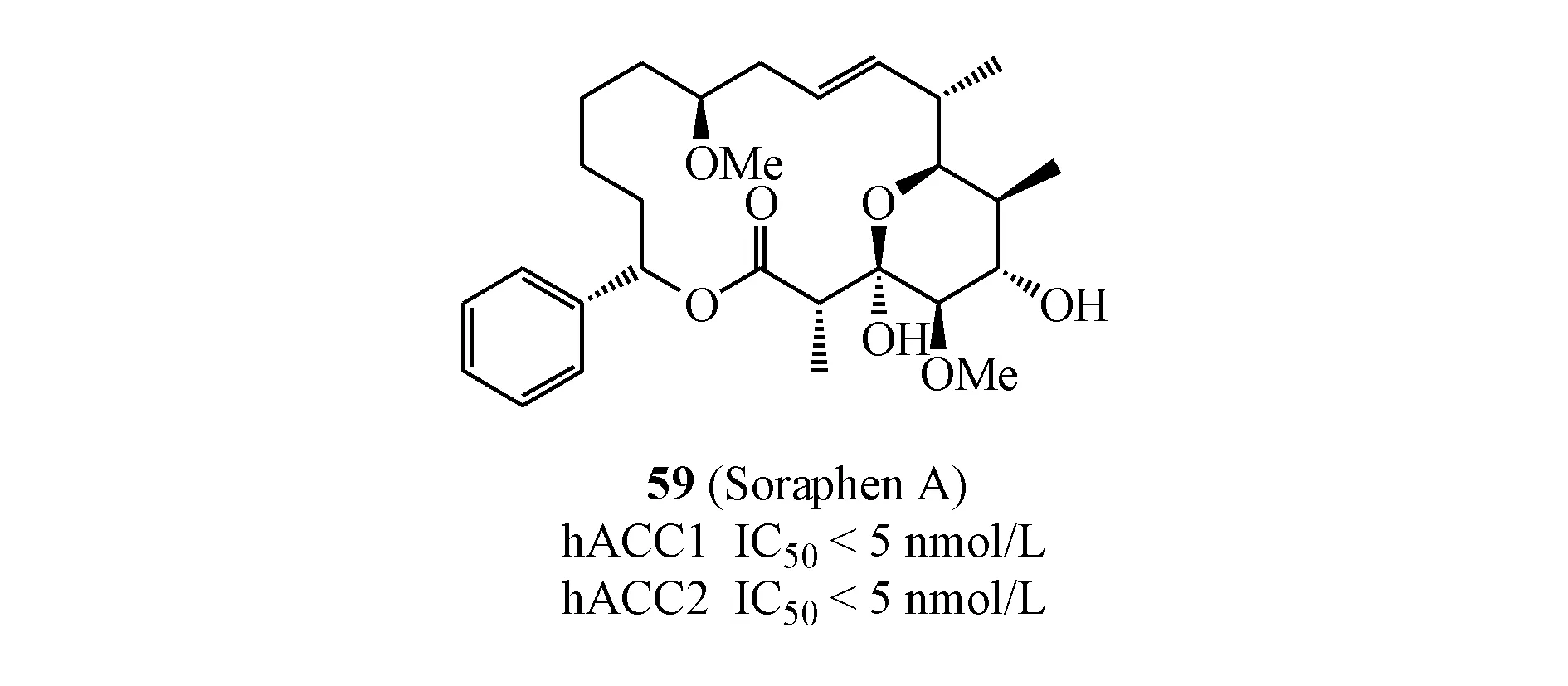
2004年,Jump等[49]报道了具有大环内酯结构的天然产物59(Soraphen A)(hACC1 IC50<5 nmol/L;hACC2 IC50<5 nmol/L)表现出较好的ACC抑制活性,其抑制机制是阻碍ACC单体的二聚化,这也是第1个明确作用于BC域的ACC抑制剂。Nimbus正是以该化合物为先导化合物进行高通量筛选,并结构优化得到ND-630。但该化合物经后续药理实验证明具有严重的生物致畸性和药物代谢性质差的缺点,随即终止研发,未见更新报道。
3 展 望
ACC作为一个传统靶点,近年来随着肿瘤、代谢性疾病研究的兴起,使该靶点获得重生。如今,全球有关ACC抑制剂的在研小分子化合物结构种类广泛而且研究机构颇多(如Pfizer、Nimbus Therapeutics以及Gilead等研发公司)。尽管目前尚无此靶点药物上市,但其作为脂肪酸合成的关键酶和限速酶,其抑制剂在治疗代谢性疾病如肥胖、2型糖尿病、非酒精性脂肪肝病、高脂血症以及肿瘤等方面发挥着重要作用。目前ND-630(GS-0976)和PF-05221304均已处于临床Ⅱ期试验研究中;且有多个小分子化合物处于生物活性测试阶段,给药效果显著。如果以ACC为靶点的抗代谢性疾病或抗肿瘤药物能够研发成功,必将会为患者带来福音。
