水稻脱落酸受体PYL2与mNeonGreen融合蛋白的原核表达及纯化
2019-06-25吴云华吴亚婷李勇
吴云华, 吴亚婷, 李勇
(中南民族大学 生命科学学院,武汉 430074)
脱落酸(ABA)在植物体内具有多种重要生理功能,如促进气孔关闭,控制种子萌发和响应外界非生物胁迫等[1],开发ABA的检测方法对研究植物生理功能及信号机制方面具有重要意义. ABA检测难点在于其在植物内含量极低和基质复杂[2],目前植物激素检测方法有气相色谱法、生物测定法、高效液相色谱法、化学发光法、质谱联用法、酶联免疫吸附法等[3], 这些方法存在样品预处理复杂或灵敏度低等问题. 因此需要开发高灵敏、原位无损检测方法.
脱落酸的信号传导是通过脱落酸与其受体识别结合实现. 研究表明:PYL蛋白家族是ABA的受体[4],水稻有12个OsPYLs同源基因[5],由于OsPYL2在所有组织中均呈高表达量[6],故本文选取水稻脱落酸受体 PYL2[Os02g0226801] 为研究对象. mNeonGreen是2013年报道的来自于一种改造文昌鱼的单体黄绿色荧光蛋白,在517 nm处有最大发射[7],光稳定性较以往报道的绿色或黄色荧光蛋白更高,可作融合标签[8].
本文将黄绿色荧光蛋白mNeonGreen融合在PYL2的C端,用原核表达系统获得融合蛋白PYL2-mNeonGreen,该融合蛋白能与ABA互作引起mNeonGreen荧光信号的改变. 基于此信号,可实现ABA的荧光检测.
1 材料与方法
1.1 材料、试剂和仪器
水稻日本晴cDNA,大肠杆菌菌株DH5α,BL21(DE3),原核表达载体pET-28a,黄绿色荧光蛋白(mNeonGreen)质粒均由中南民族大学生命科学学院生物医学分析研究组实验室保存.
内切酶NdeI、EcoR I(Takara公司);金牌MIX(擎科生物);T4 DNA连接酶(Takara);S-(+)ABA、蛋白质分子量Marker(Thermo);30%丙烯酰胺/甲叉双丙酰胺(质量比为29∶1)储液(上海生工),DNA凝胶回收试剂盒、DNA清洁回收试剂盒、质粒回收试剂盒(Axygen);DNA分子量Marker和镍亲和纯化柱(康维世纪生物);引物合成及测序(武汉擎科生物技术).
琼脂糖及聚丙烯酰胺凝胶电泳仪(DYY-6D,北京六一仪器厂);荧光光谱仪(F-2500,HITACHI);细胞破碎仪(JY96-II,杭州川一);NanoDrop One(Thermo);水浴锅(HWS-26型,上海齐欣).
1.2 载体的构建
根据重叠PCR原理,用引物设计软件Primer Premier 5.0 设计特异性引物(见表1),在PYL2和mNeonGreen蛋白编码序列之间连入一段十肽(GGGGS)2的柔性链做为融合蛋白的linker.

表1 PCR引物序列Tab.1 Primers sequences for PCR
以实验室水稻日本晴cDNA和含有mNeonGreen编码序列的质粒为模板,用PYL2和Linker-mNeonGreen的上下游引物分别扩增PYL2和mNeonGreen两个DNA片段,凝胶清洁回收后,以PYL2和mNeonGreenDNA回收产物为模板,用PYL2的上游引物和Linker-mNeonGreen的下游引物进行重叠PCR扩增出PYL2-mNeonGreen融合片段.以mNeonGreen上下游引物,PCR扩增mNeonGreen片段做实验对照.
将pET-28a质粒和扩增得到的目的片段分别用NdeI和EcoR I于37 ℃双酶切12 h,清洁回收后,4 ℃连接12 h. 连接产物转化大肠杆菌DH5α. 经过菌落PCR和双酶切鉴定阳性菌,测序验证.
1.3 蛋白表达
将pET-28a空载体质粒,pET-28a-mNeonGreen和pET-28a-PYL2-mNeonGreen重组质粒转化到大肠杆菌菌株BL21(DE3)中.从平板中挑取长势良好的单菌落[9]接种到含有卡纳霉素(50 ng/L)的LB液体培养基中,37 ℃,200 r/min,12 h培养之后按1∶100的体积比将其转接至150 mL LB培养基(50 ng/L卡纳霉素),37 ℃培养约2 h,待菌液OD值达到0.6,加入终浓度为0.8 mmol/L 的异丙基硫代半乳糖(IPTG)进行诱导,28 ℃,200 r/min,继续培养14 h.
1.4 蛋白纯化
将诱导后的150 mL mNeonGreen和PYL2-mNeonGreen菌液,放至50 mL离心管中,3000 g于4 ℃离心15 min,倒掉上清收集菌体,沉淀用30 mL 细菌蛋白抽提液(20 mmol/L Tris-HCl,500 mmol/L NaCl,pH 8.0)重悬,加入300 μL溶菌酶(10 mg/mL)和30 μL蛋白酶抑制剂. 上述菌悬液使用细胞破碎仪对细胞进行充分破碎后,10000 g于4 ℃离心15 min,收集上清,用0.22 μm过滤膜过滤,留存备用.
将提前处理好的镍柱,用ddH20冲洗3次,再用平衡缓冲液(20 mmol/L Tris-HCl,500 mmol/L NaCl,10 mmol/L 咪唑,pH 8.0)平衡2次. 将蛋白表达上清液加入镍柱中,重复3次. 先用25倍柱体积洗涤缓液(20 mmol/L Tris-HCl,500 mmol/L NaCl,40 mmol/L 咪唑,pH 8.0)冲洗镍柱,除去杂蛋白,用考马斯亮蓝检测是否除干净. 再用2倍柱体积洗脱缓冲液(20 mmol/L Tris-HCl,500 mmol/L NaCl,500 mmol/L 咪唑,pH 8.0)洗脱目的蛋白. 用考马斯检测是否洗脱干净,纯化后的蛋白用PBS透析5 h,每隔2 h换1次透析液,最后分装至200 μL EP管于-80 ℃冰箱保存.
1.5 蛋白质浓度检测和SDS-PAGE
取透析后的蛋白,用紫外分光光度法A280进行蛋白浓度测定,并对蛋白质进行SDS-PAGE分析. 取20 μL蛋白质样品,加入20 μL 2×上样缓冲液,混匀后于100 ℃加热15 min进行变性处理. 取10 μL经过处理的样品上样至SDS-PAGE凝胶中进行电泳分离. 浓缩胶电压为80 V,分离胶电压为100 V. 电泳结束后将凝胶放置平皿中加染色液染色3 h,再加入脱色液脱色至凝胶条带清晰,并于胶片观察灯上进行拍照分析.
1.6 蛋白的荧光光谱检测
用3 mL PBS缓冲液将mNeonGreen和PYL2-mNeonGreen蛋白稀释成终浓度为30 μmol/L 的溶液[6],混匀后加入到比色皿中,放入荧光分光光度计,在450 nm波长激发光下进行扫描,记录荧光信号I0. 再依次加入1 mmol/L ABA溶液2 μL,扫描记录荧光光谱Ii. 重复测定3次,取平均值I,得到荧光差值I=I0-Ii,用Origin软件作图分析,
2 结果及分析
2.1 载体构建
用上述设计引物,可分别扩增PYL2和mNeonGreen两个DNA片段(见图1),切胶回收后,取PYL2和mNeonGreen编码基因片段各1 μL做模板,PYL2的上游引物和mNeonGreen下游引物各1 μL,同16 μL金牌MIX进行重叠PCR扩增得到PYL2-mNeonGreen的DNA片段(见图1).
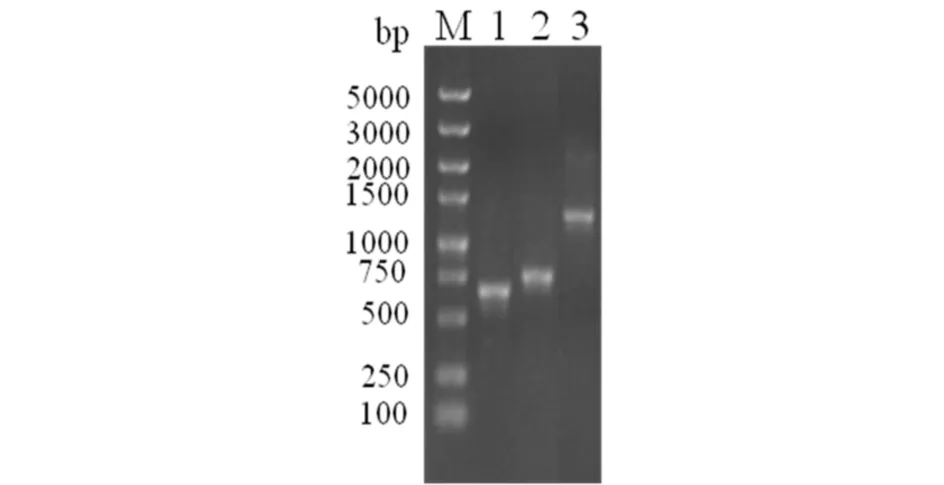
M) DL5000;1)PYL2;2) mNeonGreen;3) PYL2-mNeonGreen图1 PCR扩增PYL2, mNeonGreen和PYL2-mNeonGreen的琼脂糖凝胶电泳分析Fig.1 Agarose electrophoresis analysis of PCR products for and PYL2, mNeonGreen and PYL2-mNeonGreen
将pET28a-PYL2-mNeonGreen载体用NdeI和EcoR I进行双酶切鉴定,酶切产生大小约为1400 bp目的片段(见图2a),pET28a-mNeonGreen载体酶切产生大小约为750 bp目的片段(见图2b),均与预期大小相符. 进一步测序验证后结果表明,重组载体构建成功.
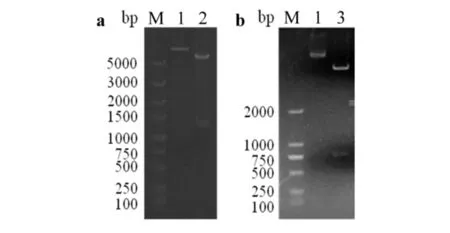
M) DL5000;1) pET-28a空载体;2)重组质粒PYL2-mNeonGreen;3) 重组质粒pET-28a-mNeonGreen图2 重组质粒pET28a-PYL2-mNeonGreen和pET28a-mNeonGreen双酶切鉴定Fig.2 Identification of recombinant plasmid pET-28a-PYL2-mNeonGreen and pET-28a-mNeonGreen by double enzyme digestion
2.2 蛋白质的表达与纯化
将pET28a-PYL2-mNeonGreen和pET28a-mNeon Green载体,转化至大肠杆菌BL21,进行诱导表达和蛋白质的分离纯化,进行SDS-PAGE检测(见图3).
PYL2-mNeonGreen融合蛋白经Ni柱亲和层析纯化和透析后在凝胶中条带清晰,在49 kDa处附近可明显看到目的蛋白条带,与理论预测49 kDa大小相符. mNeonGreen蛋白在凝胶条带中清晰且无杂带,在28 kDa处附近有目的蛋白条带,与理论预测28 kDa大小相符. 表明PYL2-mNeonGreen和mNeonGreen蛋白均获得了表达和纯化.

1)蛋白Marker;2) pET-28a空载体转化大肠杆菌BL21诱导后细胞破碎混合液;3) pET28a-PYL2-mNeonGreen载体转化大肠杆菌BL21诱导后细胞破碎混合液;4) Ni柱纯化后的PYL2-mNeonGreen蛋白表达上清样品;5) 透析后PYL2-mNeonGreen蛋白纯化样品;6) pET28a-mNeonGreen载体转化大肠菌BL21诱导后细胞破碎混合液;7) Ni柱纯化后的mNeonGreen蛋白表达上清样品;8) 透析后mNeonGreen蛋白纯化样品图3 重组蛋白表达和纯化的SDS-PAGE分析Fig.3 Characterization of the expression and purification of the recombinant proteins by SDS-PAGE
2.3 PYL2-mNeonGreen融合蛋白与脱落酸相互作用的荧光光谱检测
含有PYL2-mNeonGreen蛋白的PBS溶液,在450 nm 波长激发光下进行扫描,可产生荧光发射光谱,该光谱的最大发射波长约为520 nm,与单体黄绿色荧光蛋白mNeonGreen荧光发射光谱基本吻合[7](见图4). 依次加入终浓度为0.67, 1.33, 2.00, 2.67, 3.33 μmol/L 的ABA,其最大发射峰处的荧光强度不断降低. 以mNeonGreen蛋白为对照,如图4b所示,当依次加入等浓度ABA时,mNeonGreen的荧光强度也逐渐降低,但减弱程度明显小于PYL2-mNeonGreen蛋白荧光降低强度.

图4 脱落酸加入引起PYL2-mNeonGreen (a) 和mNeonGreen (b) 荧光光谱变化Fig.4 Fluorescence spectra changes ofPYL2- mNeonGreen (a) and mNeonGreen (b) after the addition of abscisic acid
重复检测3次取平均值,得到加入溶液浓度和荧光峰值降低程度(I0-Ii)的线性关系(见图5). 实验结果表明:随着ABA浓度的不断增加,融合蛋白PYL2-mNeonGreen的荧光强度不断降低,其降低程度大于mNeonGreen自身荧光光漂白所引起的荧光强度降低. 扣除mNeonGreen空白对照,得到ABA与融合蛋白互作引起的荧光光谱强度的净变化值(见图5内插图),此信号可用于ABA的定量检测.
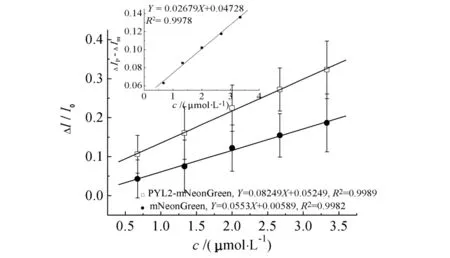
图5 PYL2-mNeonGreen和mNeonGreen蛋白的荧光峰强度与加入ABA浓度变化的线性关系Fig.5 The linear relationship between the fluorescence intensity change of PYL2-mNeonGreen or mNeonGreen and the concentration of AB
3 结语
本文采用重叠PCR技术,首次将PYL2的C端和绿色荧光蛋白mNeonGreen进行了融合,构建重组质粒pET28a-PYL2-mNeonGreen,并对该重组蛋白进行了原核表达与纯化,得到较高纯度的PYL2-mNeonGreen蛋白,在450 nm波长光激发下,该融合蛋白在518 nm处具有最大荧光发射光谱. 该最大发射峰荧光强度随ABA浓度的增加而不断降低,由于PYL2蛋白以同源二聚体存在,在结合ABA后,PYL2单体-单体相对方位的变化和二聚化界面上氨基酸的重新排布,使PYL2二聚体被削弱[10,11],引起与其融合的mNeonGreen蛋白结构改变,导致其荧光强度减弱. mNeonGreen蛋白稳定性较高,但与传统绿色荧光蛋白一样具有光漂白性[12]. 脱落酸的加入,使PYL2-mmNeonGreen的荧光强度变化值较大,此信号可于脱落酸的荧光检测.
