基于多重置换扩增技术的胃癌患者胃内微生物的宏基因组学研究
2019-06-21胡远亮程乃嘉张朝军
胡远亮,程乃嘉,张朝军,黄 云,张 炎
中国是世界上胃癌患者最多的国家。据估计,2015年中国新发胃癌679 100例,有498 000人死于胃癌[1]。众所周知,幽门螺旋杆菌(HelicobacterPylori,HP)慢性感染是胃癌发生的主要危险因素[2-3],然而感染者中,只有不到1%的人最终发展成胃癌,提示胃癌的发生、发展还与其他因素相关。DNA测序技术和生物信息学方法的发展使我们能够不依赖传统的培养手段而对人体微生物进行研究,使得大量有关人体微生物组的研究涌现[4-5]。其中,有不少研究关注人体消化道微生物在胃部疾病中的作用。多项基于16S rRNA基因测序的研究已证实胃内菌群构成复杂,除HP外还有其他多种微生物存在,并且胃内菌群失调与胃癌密切相关[6-9]。然而,对细菌16S rRNA基因的分析很难精确到种的水平,而基于全基因组测序的宏基因组学方法可以在种甚至菌株水平上分析特定微生态系统中的菌群特征,从而能够更加全面和深入地研究人体微生物和疾病的关系[4]。本研究通过多重置换扩增技术(multiple displacement amplification,MDA)与宏基因组鸟枪法测序相结合的方法研究胃癌患者和健康受试者胃内微生物的组成特征,从全基因组的角度探索胃菌群和胃癌的关系。
1 材料与方法
1.1 受试者筛选和标本采集 胃癌组(GA)纳入2017年9月—2017年12月在医院就诊并经病理学证实的胃腺癌患者,纳入者6个月内未接受抗生素治疗[10-12],4周内未使用质子泵抑制剂或其他制酸剂,未接受术前放化疗或内镜治疗;同期纳入无消化系统症状,且胃镜检查正常的来医院查体人员作为健康对照组(HC),入组标准同胃癌组。胃癌组共纳入4例,年龄(60.4±6.5)岁;男女比例3∶1;健康对照组纳入4例,年龄(53.2±5.3)岁;男女比例3∶1。本研究经医院伦理委员会审查并通过,受试者入组前签署知情同意书。受试者空腹10 h后,于早上8∶30到10∶00之间在胃镜检查室行胃镜检查。待镜头完全进入受试者胃中后,多部位吸取胃液至无菌容器,取样完毕后立即将样本置于冰上,并于0.5 h内运送至实验室处理。
1.2 标本预处理 所有步骤均在层流的生物安全柜中进行。将样本转移至50 mL离心管,用无菌0.9%氯化钠溶液稀释样本至50 mL,充分混匀后低温低速离心10 min(400 r/min,4 ℃)。保留上清,弃去含有食物残渣和人体细胞的沉淀。上清用孔径5 μm无菌滤膜过滤,保留滤液,弃去滤膜(滤膜上有低速离心未分离出的人体细胞和食物残渣),滤液再用0.22 μm滤膜过滤(此时细菌富集在0.22 μm孔径滤膜上),弃去滤液,保留滤膜。用灭菌剪刀把滤膜剪成0.5 cm×0.5 cm大小,置于无菌培养皿中,用于DNA提取。经过预处理,标本中的人体细胞和食物残渣等得到有效去除,将大大提高细菌DNA提取的质量,降低测序成本,并提高数据分析的准确性。
1.3 DNA提取 采用MO-BIO PowerSoil DNA Isolation Kit对预处理后的滤膜碎片进行DNA提取,该方法把化学裂解和珠磨结合起来,保证了细菌DNA提取效率。
1.4 DNA扩增 采用MDA方法对提取DNA进行扩增,扩增反应时间为6 h。
1.5 构建测序文库 用0.9倍DNA溶液体积的AMPure XP beads纯化扩增产物。使用Covaris S2超声破碎仪打断纯化后的DNA扩增产物,选取程序DNA 300超声破碎。片段化的DNA经过0.9倍DNA体积的AMPure XP beads纯化后,使用NEBNext ultra DNA library prep kit for Illumina试剂盒构建DNA文库。
1.6 文库质检 建库完毕后即送到北京大学生物光学动态成像中心的测序平台进行质检分析。
1.7 高通量测序 文库质检完毕,即送到诺禾致源公司,使用Illumina HiSeq Xten测序平台进行双端PE150测序。
1.8 数据质控 使用fastqc对测序原始数据进行质量评估,用Trimmomatic[13]去接头和低质量序列,并结合bowtie2[14]比对工具去除原始数据中的宿主序列,保留高质量的微生物DNA序列。
1.9 统计学处理 使用MetaPhlAn2[15]对质控后的数据进行物种注释,得到每个样本的物种类别及物种相对丰度。应用SPSS 22.0统计软件,用R语言对数据进行可视化。组间菌种数目比较采用Student’s t-test。使用STAM[16]软件分析组间差异物种。比较组间优势物种相对丰度采用Mann-Whitney U test。以P<0.05为差异比较具有统计学意义。
2 结果
2.1 MDA能显著放大提取的微量胃液微生物DNA 各样本提取的微生物DNA总量分布于1~5 ng,难以达到构建高质量宏基因组测序文库的要求。通过MDA扩增,各个样本的DNA显著放大(207.27±33.17)倍,可以直接用于全基因组文库的构建。为了提高扩增效果的可比性,每个DNA样本的扩增起始量均为1 ng(表1)。
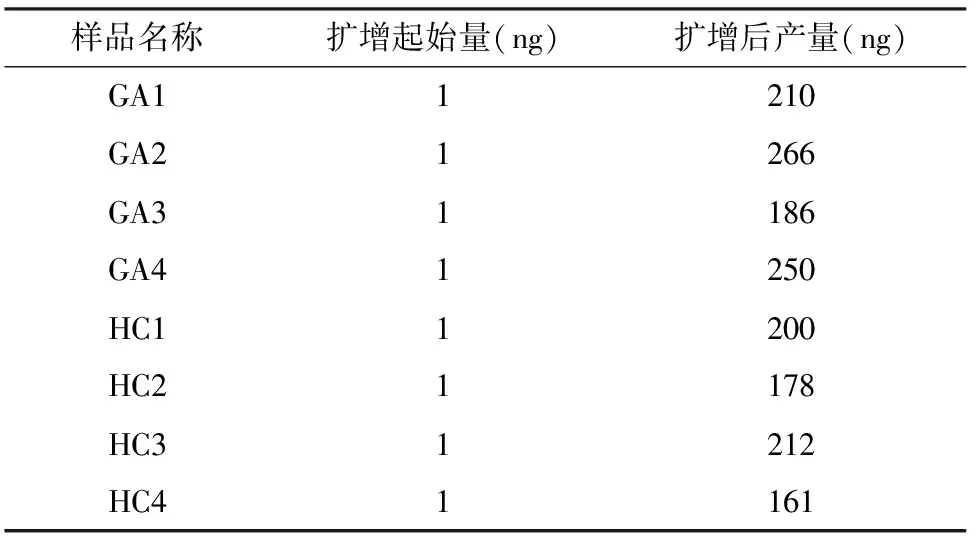
表1 MDA能显著扩增各样本的微量微生物DNA
注:GA1-4和HC1-4分别代表胃癌组和健康对照组样本
2.2 测序数据质量评估 样本的数据量、Q20值、Q30值、测序错误率以及GC含量相关数据具体见表2。所有样本测序平均通量为3.28G,Q30值均大于91%,测序错误率均低于0.02%,可见测序质量较好。
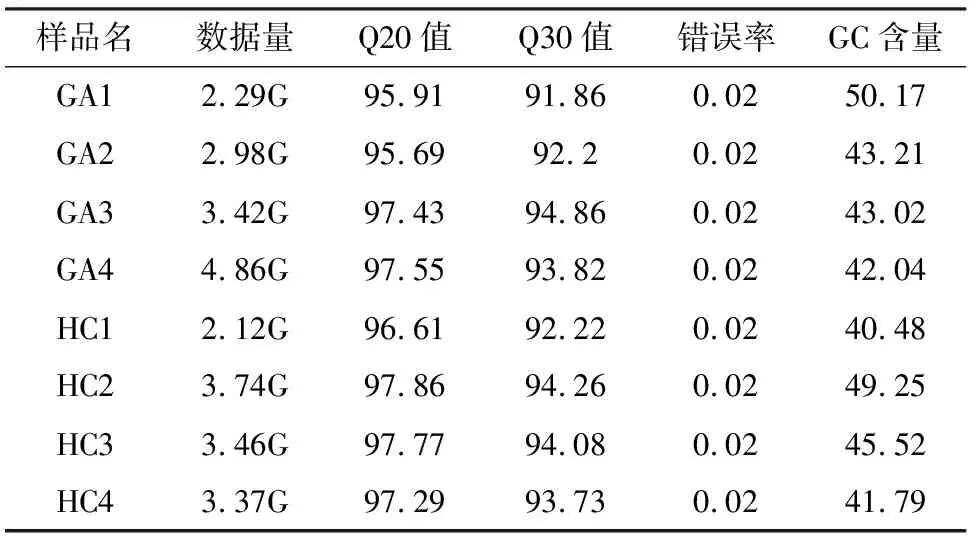
表2 全基因组测序数据质量评价
2.3 组间微生物组成的整体分布 经测序数据质控,去除低质量序列和宿主序列,得到高质量的微生物DNA序列。用MetaPhlAn2对质控后的数据进行物种注释,得到各样本物种组成及物种相对丰度。所有样本共鉴定出131个菌种,胃癌患者胃内菌群多样性明显低于健康受试者(Student’s t-test,P<0.01,t=4.189,图1)。在细菌门水平,受试者胃液菌群主要由变形菌门、拟杆菌门、厚壁菌门、梭杆菌门组成(图2)。同时,HC组中HP的平均相对丰度高于GA组(14.19%±22.25% vs 32.05%±28.24%),但差异比较无统计学意义(Mann-Whitney U test,P=0.383,Z=0.871)。
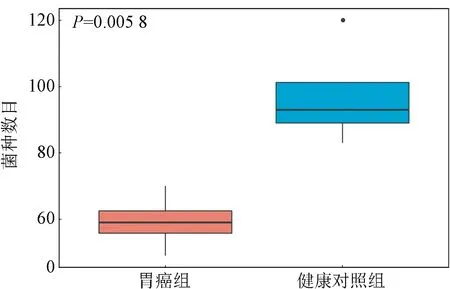
图1 胃内菌群多样性(菌种数目)比较
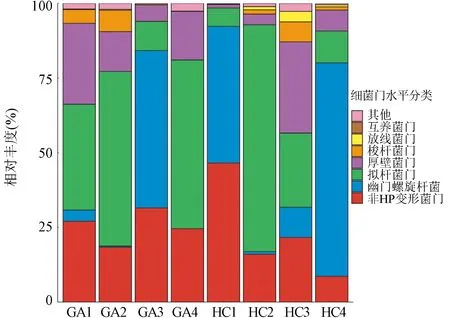
图2 细菌门分类水平各样本的物种相对丰度堆叠柱状图
2.4 组间显著差异的菌种 为了比较组间优势物种的差异,我们筛选出各个样本中相对丰度大于1%的优势物种,菌种相对丰度(%)经Log2变换,使用STAMP软件对组间优势物种进行分析。我们发现牙髓卟啉单胞菌、口腔链球菌、干燥奈瑟菌在GA组中的相对丰度明显高于HC组(Mann-Whitney U test,P<0.05,W=10.0,图3)。
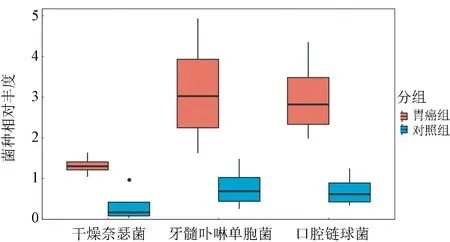
图3 组间显著差异的菌种
3 讨论
DNA测序技术的发展使我们能不依赖于传统的培养手段而对人体内大量无法培养的微生物进行研究,不断更新着我们对人体微生物的认识[4]。人体内细菌的数目大约与人体细胞的数目相当,且主要集中在消化道[17]。它们与人类共进化,在人体的生长发育、稳态维持和疾病的发生、发展中都扮演着至关重要的角色,包括癌症[18-20]。其中,HP感染已被证实是患胃癌的主要危险因素[2-3]。同时,多项研究也表明除HP外的胃内其他细菌也和胃癌的发生、发展相关[6-7,9]。然而以往的研究大多基于对细菌16S rRNA基因的分析,使用全基因组鸟枪法测序对胃癌患者胃内菌群的研究微乎其微[21]。本研究使用MDA技术扩增从胃液中提取的微量细菌DNA,并用扩增产物构建了高质量的测序文库,用于高通量测序,得到了高质量的测序数据,并对数据进行生物信息学分析,从很大程度上证实了全基因组扩增的方法用于人体胃内微生物组研究的可行性。
以往的研究主要把MDA用于扩增真核细胞基因组,特别是用于单细胞测序[22-23]。也有不少学者把MDA用于扩增成分已知的微生物基因组,并对其扩增的覆盖度和均匀度进行评估[24-25]。还有一些研究把细菌分选技术与MDA技术结合起来,以求得到某种或某些细菌的全部基因组序列[26-27]。这些研究都证明了MDA用于扩增DNA的可靠性和有效性。然而,很少有学者将MDA用于扩增复杂微生物样品中的DNA。虽然我们未进一步对扩增覆盖度和均匀度进行验证,但初步的分析结果证实通过该方法可以从微量的DNA中检测到大量精确定位到种的微生物,证明了该方法用于胃内微量微生物检测的可行性。
不少基于对胃黏膜细菌16S rRNA基因的研究发现,胃癌患者的HP感染率和/或HP相对丰度要低于正常人和/或浅表性胃炎患者[7,28]。可能原因是HP慢性感染导致胃酸分泌减少,从而使其他本不适应酸性环境的细菌能够在胃内更好存活,而又反过来影响HP自身在胃内的定植[29-30]。本研究收集的8个样本中,4名健康人的胃液均检测到不同丰度的HP序列,而4名胃癌患者有3名存在HP感染,且健康对照组中HP的平均相对丰度高于胃癌组,从一定程度上反映了胃癌患者HP感染率下降的趋势,与之前的研究相符。然而,HP序列相对丰度在8名受试者中差异较大(23.12±26.95),提示HP携带者个体之间感染程度差异较大。在抗生素耐药越来越严重的今天,这种个体之间感染程度的巨大差异也许对HP感染控制策略的个体化制定有一定指导意义。
我们从所有样本中一共鉴定出131种细菌,并发现与健康对照组比较,牙髓卟啉单胞菌、口腔链球菌、干燥奈瑟菌在胃癌组中富集。牙髓卟啉单胞菌和口腔链球菌是人体口腔内的共生菌,而干燥奈瑟菌主要定植于上呼吸道黏膜,它们在胃癌受试者胃液中大量存在,也提示胃癌患者胃内微环境的变化可能允许来自身体其他部位的特定细菌在胃内定植,但这种定植与胃部疾病的因果关系有待进一步研究。我们还发现胃癌患者胃液内菌种多样性显著低于健康对照组,这与之前对胃黏膜菌群的研究一致[6-7]。本研究样本量较少,尽管样本采集和数据分析均采用统一的方法,但只有通过更大样本量的研究和多人群的验证才能进一步排除混杂和随机因素,得到更加肯定的结论。
总之,我们首次把MDA和宏基因组鸟枪法测序结合起来研究胃癌患者胃内菌群的特征,证实了该方法的可行性。该方法可用于从菌种甚至菌株的水平上研究与胃癌发生、发展相关的微生物,并可用于微量病原体的检测。同时,我们发现特定菌种在胃癌患者胃液内富集,提示其作为胃癌生物学标志物的潜在价值。在以后的研究中纳入更多受试者,涉及更广的人群,进一步证实该方法的可靠性,并探索菌群与胃癌发生、发展的因果关系。
