磁性核壳Fe3O4/P(GMA-DVB)-SH-Au复合催化剂的制备及催化性能
2019-06-19马明亮杨玉莹贾新城孔令运池丽凤
马明亮,杨玉莹,吕 平,贾 丽,贾新城,陈 柳,孔令运,池丽凤
(青岛理工大学 土木工程学院,山东 青岛 266033)
随着水污染问题日益严重,有机污染物因毒性大含量多且自身难降解,而成为主要的水污染来源,4-硝基苯酚(4-NP)就是其中之一[1-4]。近年来,许多生物和物理化学方法被用来处理有机水污染,但是常规生物处理由于某些不可生物降解物质的存在而受到阻碍,同样,物理化学技术如膜分离、汽提、混凝等,通常在初始生产阶段、能源消耗或化学品使用等方面花费昂贵,成本较高[5]。因此,开发一种用于降解有机污染物的高效环保且经济的方法具有重要意义。在现代化学研究中,利用催化剂及催化反应解决水污染问题已经引起了人们的广泛关注[6-8]。Au纳米粒子(NPs)因具有不同于块状金属的独特性质,如优异的选择性和良好的反应性,在硼氢化物的存在下可以高效催化还原4-NP,且不产生二次污染,已被作为催化剂广泛应用于各催化反应[9-12]。
近年来,虽然关于纳米催化剂的研究取得了一定进展,但仍存在一些问题。一方面,为了避免AuNPs团聚而使其具有更好的分散性和更高的催化活性,需要引入合适的载体材料。另一方面,从催化体系的混合溶液中分离和回收纳米催化剂是非常困难的,催化剂的回收和再利用仍然是一个很大的挑战[13]。由于Fe3O4纳米粒子具有优异的磁性能和化学稳定性,因此可作为AuNPs的载体材料,以便对纳米Au催化剂进行磁回收[14-16]。Zhao等[17]将Fe3O4纳米粒子引入其设计中,极大地促进了材料的回收。尽管Fe3O4纳米粒子可以直接作为载体来负载活性粒子,但无法保证结构的稳定性,活性粒子的脱落会导致复合材料催化性能的降低。因此,使用易于进行表面改性的有机聚合物层对Fe3O4纳米粒子进行包覆,利用表面官能团来固定及分散活性粒子,是设计既具有磁性又高效稳定的催化剂的思路之一。
基于此,本实验首先通过水热法合成空心Fe3O4磁球,然后通过硅烷偶联剂KH570对Fe3O4磁球进行表面处理,随后在一定的条件下通过蒸馏沉淀聚合使P(GMA-DVB)聚合物层包覆在Fe3O4磁球外表面得到Fe3O4/P(GMA-DVB)核壳结构。壳层P(GMA-DVB)一方面可以起到保护磁核免受底物腐蚀的影响,另一方面,相较于无机物壳层,有机层更易进行官能团改性,方便引入巯基基团(—SH)改善活性粒子的分散及稳定性。
1 实验材料与方法
1.1 试剂
六水合三氯化铁(FeCl3·6H2O),聚丙烯酸钠,尿素,柠檬酸钠,无水乙醇,硅烷偶联剂KH570,氨水(NH3·H2O),乙腈,偶氮二异丁腈(AIBN),无水碳酸钠(Na2CO3),氯金酸,磷酸二氢钠,磷酸氢二钠,4-硝基苯酚(4-NP)和硼氢化钠(NaBH4)购自上海埃彼化学试剂有限公司。甲基丙烯酸缩水甘油酯(GMA),二乙烯基苯(DVB),二硫苏糖醇(DTT)购自阿拉丁试剂(上海)有限公司。胱氨二盐酸盐购自成都贝斯特试剂有限公司。所有试剂都是分析级并且不经进一步纯化而使用。
1.2 空心Fe3O4微球的制备
按照文献报道的水热法合成单分散中空Fe3O4微球[18]。称取0.60g聚丙烯酸钠溶于80mL去离子水中,待完全溶解后,称取1.35g FeCl3·6H2O、1.00g尿素、2.94g柠檬酸钠溶于聚丙烯酸钠溶液,待完全溶解后,将上述溶液转移至100mL聚四氟乙烯衬套的不锈钢反应釜中,220℃反应20h,冷却至室温后取出。
1.3 核壳结构Fe3O4/P(GMA-DVB)微球的制备
取制得的0.30g Fe3O4微球与160mL无水乙醇和40mL去离子水混合,随后分别加入4mL KH570和4mL NH3·H2O,在40℃下快速搅拌24h,得到表面带有反应性双键的Fe3O4微球。
取0.20g修饰后的Fe3O4微球与80mL乙腈混合。然后,加入2.00g AIBN,1.00g GMA单体和1.00g DVB单体。反应体系水浴加热到90℃,当馏出物达到40mL时停止反应,得到核壳结构Fe3O4/P(GMA-DVB)微球。
1.4 Fe3O4/P(GMA-DVB)-SH-Au微球的制备
取0.15g的Fe3O4/P(GMA-DVB)微球溶于30mL 0.50mol/L的Na2CO3溶液中,超声分散均匀后,将230.50mg的胱氨二盐酸盐加入到上述体系中,在45℃恒温水浴环境下搅拌反应12h。分离洗净之后,将微球在pH值为7.8的环境下加入到10mL 12.4mg/mL的DTT溶液中,25℃搅拌2h,得到Fe3O4/P(GMA-DVB)-SH微球。
取0.1g Fe3O4/P(GMA-DVB)-SH微球,加入到20mL去离子水中,并滴加3mL 10mg/mL的氯金酸溶液,25℃下搅拌反应30min。之后,在上述体系中加入1mL 0.60mol/L的NaBH4,继续反应3h,得到Fe3O4/P(GMA -DVB)-SH-Au催化剂。上述产物都利用外部磁铁进行磁分离,并用无水乙醇与去离子水洗涤,冷冻干燥。
1.5 仪器与分析表征
通过使用JEM-3010透射电子显微镜(TEM)和JSM-6700F扫描电子显微镜(SEM)获得样品的形貌。用Bruker Tensor 27光谱仪记录傅里叶变换红外光谱(FTIR)。用Shimadzu XRD-7000 X射线衍射仪(XRD)表征样品的晶态结构。用SDT Q600热重分析仪(TGA)测定样品的热性能。用LakeShore7307振动样品磁强计(VSM)研究样品的磁性能。用UV-5200PC紫外可见分光光度计(UV-vis)监测样品的催化性能。
1.6 催化实验
取1mL 0.6×10-3mol/L的4-NP溶液与2mL 0.1mol/L的冰冷新制的NaBH4水溶液混合均匀,然后将1mL 0.1mg/mL的Fe3O4/P(GMA-DVB)-SH-Au悬浮液加入到上述溶液中,每隔一段时间用UV-vis监测反应,得到催化过程中的吸光度曲线。在磁分离循环使用实验中,Fe3O4/P(GMA-DVB)-SH-Au催化剂通过磁铁进行磁分离,用去离子水冲洗后重复使用。
磁性核壳结构Fe3O4/P(GMA-DVB)-SH-Au催化剂的合成如图1所示。

图1 Fe3O4/P(GMA-DVB)-SH-Au核壳磁性催化剂的合成Fig.1 Synthesis process for cole-shell Fe3O4/P(GMA-DVB)-SH-Au magnetic catalyst
2 结果与分析
2.1 Fe3O4/P(GMA-DVB)-SH-Au催化剂的表征
2.1.1 TEM/SEM分析
图2为Fe3O4/P(GMA-DVB)-SH-Au催化剂的结构与形貌图。图2(a)为水热法制备的空心Fe3O4微球的TEM照片,从TEM图像看,制备的微球的外围颜色较深,内部较浅,表明磁球是空心球形结构,粒径均匀约为180nm。空心Fe3O4微球与实心球相比具有密度较低,溶液分散性好的特点。图2(b)为Fe3O4/P(GMA-DVB)微球的TEM照片,可以清楚地看到,Fe3O4表面具有厚度约80nm的壳,这证明通过蒸馏沉淀聚合法成功合成了P(GMA-DVB)聚合物并且将其包覆在空心Fe3O4磁核的外表面上,核壳结构制备成功。图2(c)与2(d)分别为Fe3O4/P(GMA- DVB)-SH-Au催化剂的TEM与SEM照片,可以看出粒径约为8nm的Au纳米粒子被吸附并分散在载体表面。TEM和SEM谱图直观地说明制备出核壳结构Au负载催化剂。

图2 材料的微观形貌图 (a)Fe3O4微球TEM照片;(b)Fe3O4/P(GMA-DVB)微球TEM照片;(c)Fe3O4/P(GMA-DVB)-SH-Au微球TEM照片;(d)Fe3O4/P(GMA-DVB)-SH-Au微球SEM照片Fig.2 Microscopic topographies of materials (a)TEM image of hollow Fe3O4 microspheres;(b)TEM image of Fe3O4/P(GMA-DVB) microspheres;and (c) TEM image of Fe3O4/P(GMA-DVB)-SH-Au microspheres;(d)SEM image of Fe3O4/P(GMA-DVB)-SH-Au microspheres
2.1.2 FTIR/XRD分析
图3为制备样品的FTIR光谱图,曲线a中580cm-1处为Fe3O4微球上Fe—O键的红外特征吸收峰[19],曲线b中1727cm-1为GMA骨架上的酯羰基振动峰,1191cm-1处为C—O—C基团,908cm-1处为GMA骨架上的环氧基红外特征峰,844cm-1处归属为DVB上苯环的红外吸收峰,说明空心Fe3O4微球上成功包覆了聚合物P(GMA-DVB)。曲线c为Fe3O4/P(GMA-DVB)-SH样品的红外光谱图,可以看到在2556cm-1处出现了一个红外吸收峰,这是—SH基团的红外特征峰,而908cm-1处的环氧峰消失,说明环氧开环接上了—SH基团,成功制备了Fe3O4/P(GMA-DVB)-SH微球。在曲线d中,由于—SH与Au的配位作用,2556cm-1处吸收峰消失。
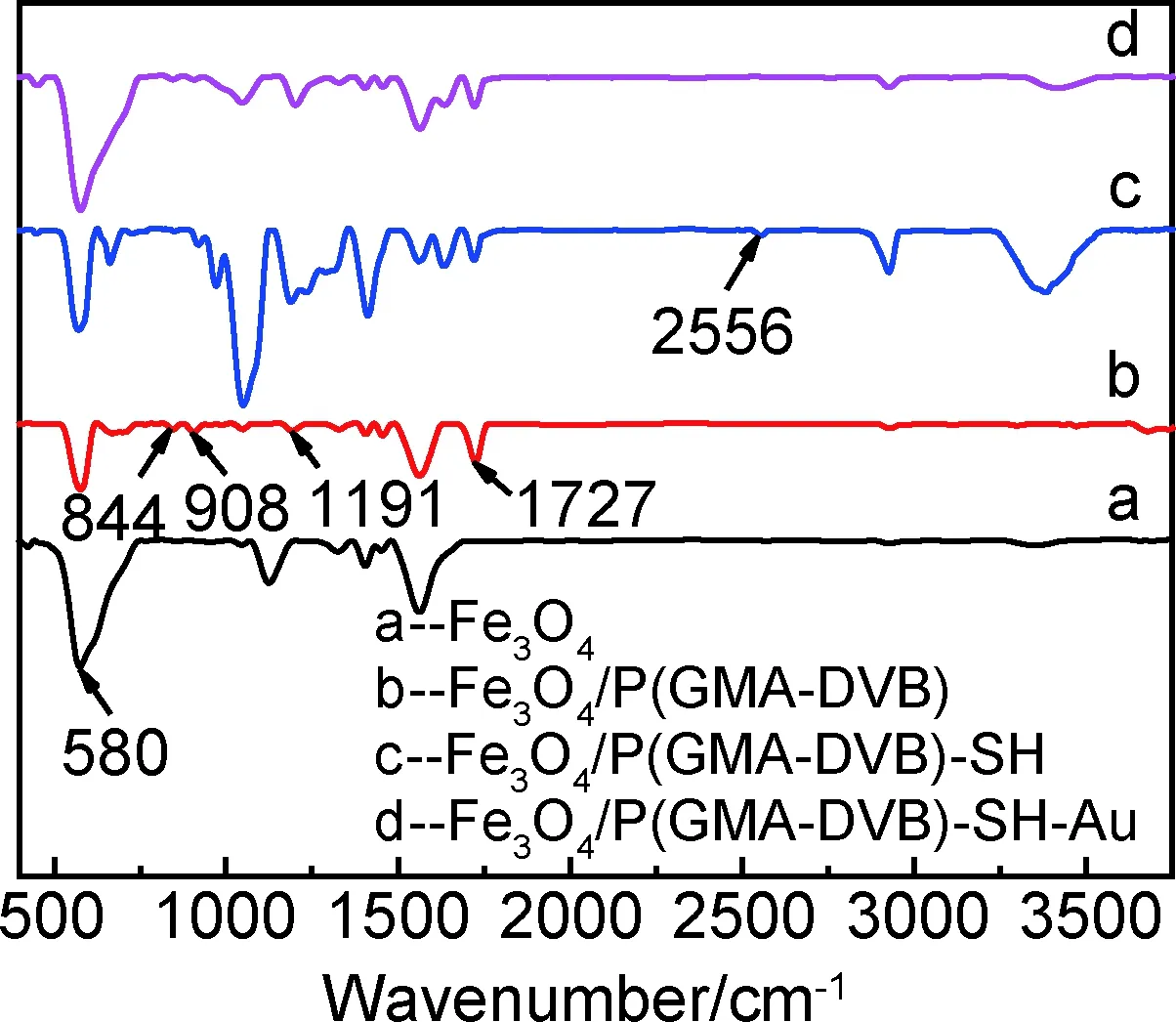
图3 微球的FTIR光谱Fig.3 FTIR spectra of microspheres

图4 微球的XRD谱图Fig.4 XRD spectra of microspheres
图4为Fe3O4微球和磁性Fe3O4/P(GMA-DVB)-SH-Au催化剂的XRD图。曲线a中,在2θ为30.4°,35.4°,43.2°,53.4°,57.2°和62.7°处分别存在6个衍射峰,分别对应于Fe3O4晶体的(220),(311),(400),(422),(511)和(440)晶面,与Fe3O4的标准PDF卡片(JCPDS 85-1436)一致[20-21]。曲线b是吸附了Au粒子的最终催化剂样品的XRD图,在38.0°,44.0°,64.0°和78.0°处出现了4个与Au晶体(111),(200),(220)和(311)晶面相对应的衍射峰[22],表明Au3+被成功还原为Au原子,并且Fe3O4微球与Au粒子的衍射峰峰型尖锐,晶型良好。
2.1.3 TGA/VSM分析
通过TGA分析进一步研究了核壳结构中聚合物涂层的包覆情况(图5)。从曲线a可以看出,当升温到100℃左右时由于Fe3O4样品中物理吸附水的蒸发,其质量轻微下降0.15%(质量分数,下同)。与曲线a类似,曲线b中Fe3O4/P(GMA-DVB)-SH-Au样品在100℃左右存在轻微的质量损失(约0.2%)。空心Fe3O4微球在250~350℃范围内产生约3.1%的质量损失,这主要是由于制备空心Fe3O4微球时残余有机物在高温下被分解,之后升高温度质量保持不变。Fe3O4/P(GMA-DVB)-SH-Au样品在230~470℃范围内的失重量为45%,主要原因是随着温度升高Fe3O4/P(GMA-DVB)-SH-Au表面聚合物被分解,重量下降,470℃后聚合物分解完全,质量保持不变。上述分析也可表明,Fe3O4/P(GMA-DVB)-SH-Au催化剂在200℃以下具有较高的热稳定性。
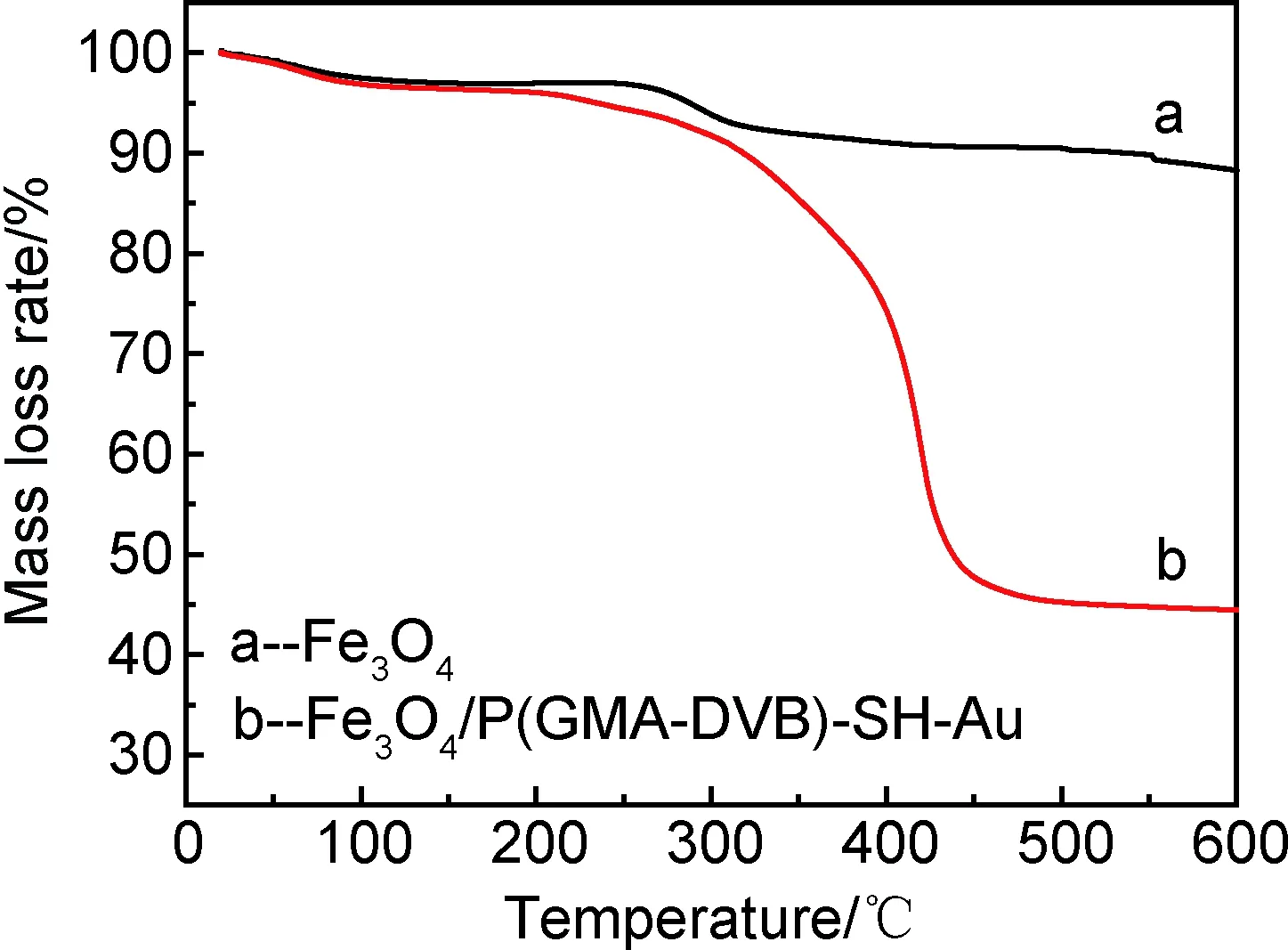
图5 微球的TGA曲线Fig.5 TGA curves of microspheres
作为可回收磁性催化剂,样品的磁性能至关重要。为了评价Fe3O4/P(GMA-DVB)-SH-Au催化剂的磁性能,通过VSM分别测试了Fe3O4微球和Fe3O4/P(GMA-DVB)-SH-Au样品的磁饱和强度。图6中空心Fe3O4微球的磁饱和度约为72.90A·m2·kg-1,包覆聚合物的Fe3O4/P(GMA-DVB)-SH-Au微球的磁饱和度约为38.37A·m2·kg-1。从图7宏观磁性分离实验中可看出,将外部磁铁放置在含有Fe3O4/P(GMA-DVB)-SH-Au催化剂悬浮液的小瓶旁边时,催化剂可以在磁场的作用下从溶液中被快速分离出来,表明所制备的磁性催化剂能够满足磁分离的要求,可以实现催化剂材料与混合溶液的高效分离回收。

图6 微球的VSM曲线Fig.6 VSM curves of microspheres

图7 宏观磁性分离实验 (a)Fe3O4/P(GMA-DVB)-SH-Au分散在水中;(b)外加磁场对微球的吸引Fig.7 Macroscopic magnetic separation test(a)Fe3O4/P(GMA-DVB)-SH-Au dispersed in water,(b)external magnetic field attraction of microspheres
2.2 Fe3O4/P(GMA-DVB)-SH-Au催化剂的催化性能
2.2.1 催化反应活性分析
为了评价Fe3O4/P(GMA-DVB)-SH-Au催化剂在催化还原4-NP反应中的催化活性,分别取适量的NaBH4溶液、4-NP溶液及Fe3O4/P(GMA-DVB)-SH-Au悬浮液混合,一定的时间间隔后利用UV-vis监测吸光度变化,结果如图8所示。一般而言,由于添加NaBH4时形成了4-硝基苯酚离子,因此4-NP溶液的紫外吸收峰将从317nm红移至400nm处[23]。因此,通过用UV-vis测量400nm处硝基化合物的吸光度变化来监测催化反应的进程。在4-NP与NaBH4反应体系中加入Fe3O4/P(GMA-DVB)-SH-Au催化剂后,图8中可看到400nm处的峰值出现持续降低,300nm处对应4-氨基苯酚(4-AP)的吸收峰值不断升高,表明催化还原4-NP为4-AP的反应正在进行,10min后反应完成。由于反应体系中NaBH4的浓度远大于4-NP的浓度,所以催化反应的速率常数k可以通过4-NP的拟一级动力学来计算[24]。如图9所示,得到ln(Ct/C0)与催化时间t的线性关系。使用公式ln(Ct/C0)=-kt来计算,其中Ct和C0分别是时间t和0时的4-NP浓度[25]。可以计算出k=0.006s-1,表明磁性核壳结构Fe3O4/P(GMA-DVB)-SH-Au催化剂对4-NP的还原效率较高。
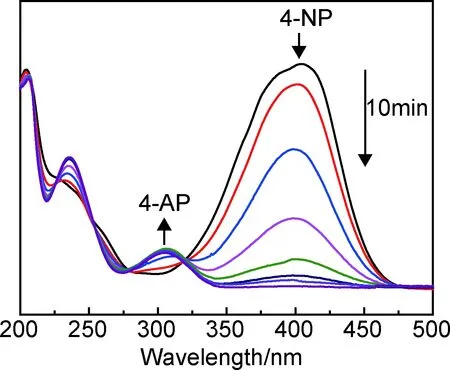
图8 催化反应的UV-vis光谱Fig.8 UV-vis spectra of the catalytic reaction
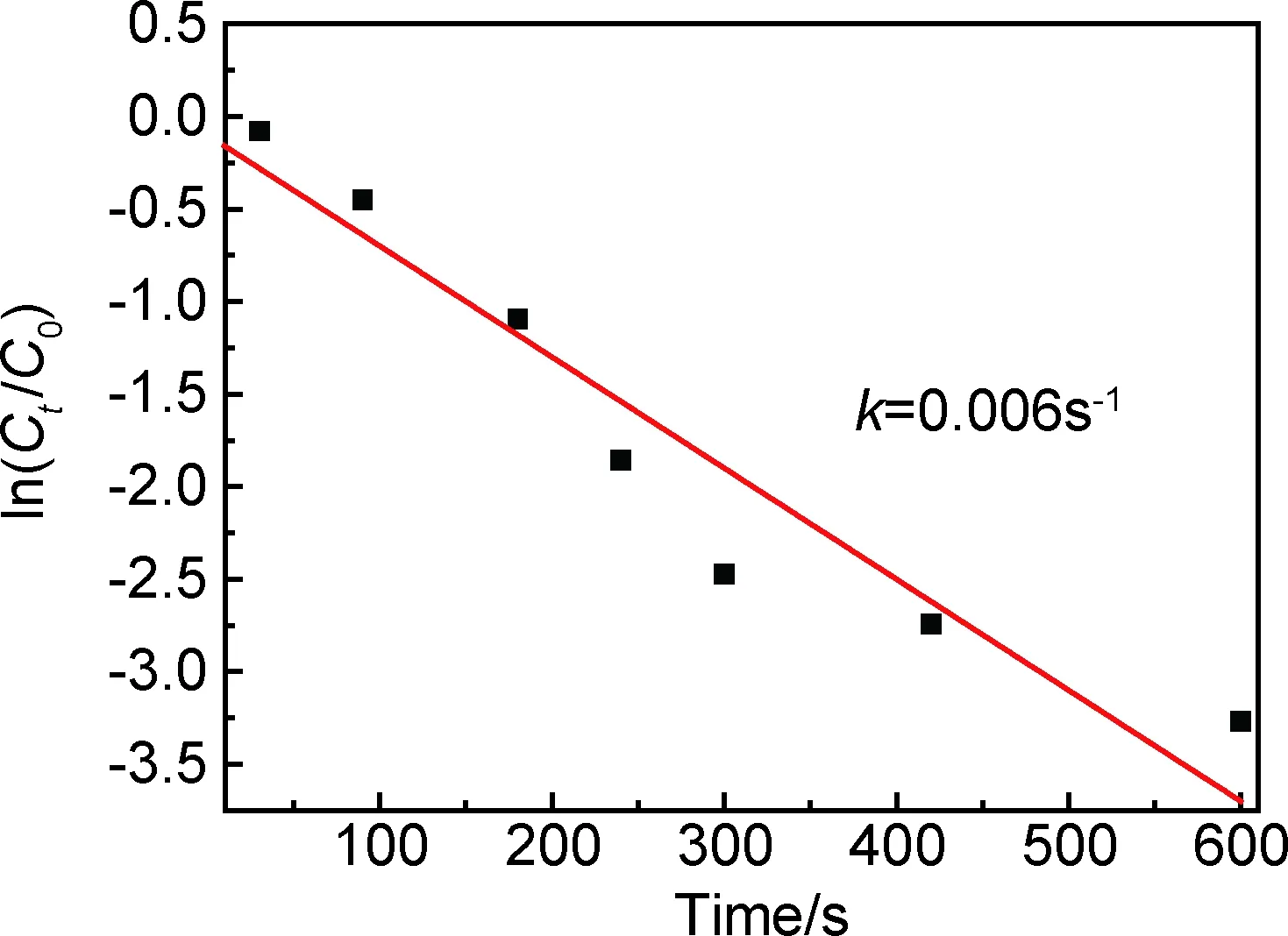
图9 ln(Ct/C0)与催化时间的关系图Fig.9 Plot of ln (Ct/C0) vs catalytic time
为了进一步考察Fe3O4/P(GMA-DVB)-SH-Au催化剂的催化性能,记录了不同条件下4-NP的降解情况,如表1所示。当反应体系中仅有4-NP和NaBH4时,4-NP不会被显著降解,转化率仅有4.49%。Fe3O4/P(GMA-DVB)-SH样品进行等条件测试,10 min后转化率为5.03%,与4.49%结果相近,转化率略高可归因于材料对4-NP分子的吸附作用。在反应体系中加入Fe3O4/P(GMA-DVB)-SH-Au催化剂,10min反应完成,4-NP的转化率高达96.26%,对比分析可知,Fe3O4/P(GMA-DVB)-SH-Au催化剂具有优异的催化性能。

表1 不同条件下4-NP的降解Table 1 Degradation of 4-NP with different conditions
2.2.2 催化循环稳定性分析
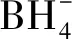

图10 Fe3O4/P(GMA-DVB)-SH-Au催化的循环实验结果Fig.10 Recycle results of reactions catalyzed by Fe3O4/P(GMA-DVB)-SH-Au
3 结论
(1)通过蒸馏沉淀聚合将P(GMA-DVB)包覆到Fe3O4磁球表面,引入—SH基团之后吸附Au粒子,制备出的样品包覆效果良好,核壳结构明显,Fe3O4磁核粒径均匀约为180nm,聚合物壳层厚度约80nm。Fe3O4/P(GMA-DVB)核壳结构负载有约8nm粒径的Au粒子,成功合成了Fe3O4/P(GMA-DVB)-SH-Au催化剂。
(2)在催化还原4-NP反应中,所制备的催化剂显示出优异的催化效率(96.26%),经8次循环使用后催化效率仍能保持80%以上。Fe3O4/P(GMA-DVB)-SH-Au催化剂既可高效催化降解催化4-NP,又可回收循环利用,具有良好的应用前景。
