胃康散质量标准研究
2019-06-19庄伟香黄木土
庄伟香,李 辉,黄木土,李 冬,刘 珊,李 健
(1.中国人民解放军南部战区总医院药剂科;2.广州市老年慢病患者合理用药重点实验室,广东 广州 510010)
胃溃疡是临床常见的消化系统疾病,致病因素较多,病理机制迄今尚未完全清楚[1-2],近年消化性溃疡的发病率逐年增长。中医药在治疗胃溃疡方面有渊源深厚的理论基础和大量丰富的临床实践经验,可发挥中药多成分多靶点的作用优势[3]。胃康散在我院一五七临床部临床应用多年,疗效显著,是由海螵蛸、白矾、白及、钟乳石(煅)、两面针、延胡索、甘草等药材组成,出自《广州部队医院制剂选编(1989)》,又名复方入地金牛散,主要用于制酸止痛,止血生肌,是治疗活动性胃溃疡的中药制剂。处方中海螵蛸为君药;白及为臣药;两面针、延胡索为佐药,甘草为使药。原质量标准中仅包括显微鉴别法和化学反应法,处方中碳酸钙等其他矿物质成分较多,不能追踪该成分从药材到制剂的转移过程,而氯化两面针碱仅存于两面针中,其转移过程易追踪。因此,本实验拟建新的质量控制方法,对处方中白及、延胡索、甘草进行薄层色谱(TLC)定性鉴别,采用高效液相色谱法(HPLC)测定处方中氯化两面针碱的含量,为其进一步开发奠定基础。
1 仪器与试药
1.1 仪器
高效液相色谱仪(Agilent 1260系列,包括二元泵、自动进样装置、DAD检测器、柱温箱等),(美国安捷伦科技有限公司);SHZ-D(Ⅲ)水循环式真空泵(巩义市予华仪器有限责任公司);HWS-28型恒温电热水浴锅(上海一恒科学仪器有限公司);KP03型超声仪(广州市科普超声电子技术有限公司)。
1.2 药品与试剂
胃康散(中国人民解放军南部战区总医院自制制剂,批号:171130,170331,180424。规格:3 g);白及对照药材(批号:121262-201405),甘草对照药材(批号:120904-201519),延胡索乙素对照品(批号:120928-201006),氯化两面针碱对照品(批号:110848-200603)(中国食品药品检定研究院);硅胶G薄层板(美国默克公司);水为两重蒸馏水,甲醇为色谱纯,其他试剂为分析纯。
2 方法与结果
2.1 定性分析
2.1.1 延胡索的鉴别 取胃康散3 g,加甲醇50 ml,超声处理30 min,滤过,滤液蒸干,残渣加水10 ml溶解,加浓氨试液调至pH 11~12[4-5],用乙醚振摇提取3次,每次10 ml,合并乙醚液,蒸干,残渣加甲醇1 ml使溶解,作为供试品溶液[6]。另取延胡索乙素对照品适量,加适当甲醇制成1 mg/ml的对照品溶液。再按制备工艺,配制不含延胡索的阴性样品,同供试品溶液配制方法制成阴性对照液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验[7],吸取样品溶液和阴性对照液各10 µl,对照品溶液2 µl,分别依次点于同一硅胶G薄层板上,以正己烷-三氯甲烷-甲醇(7.5:3.5:1)为展开剂(充分预饱和),室温展开,上展约8 cm处,取出。晾干后置碘缸熏至斑点显色清晰,约6 h后挥去板上吸附的碘后,置紫外灯(365 nm)下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同绿色荧光主斑点。阴性对照无干扰。见图1。
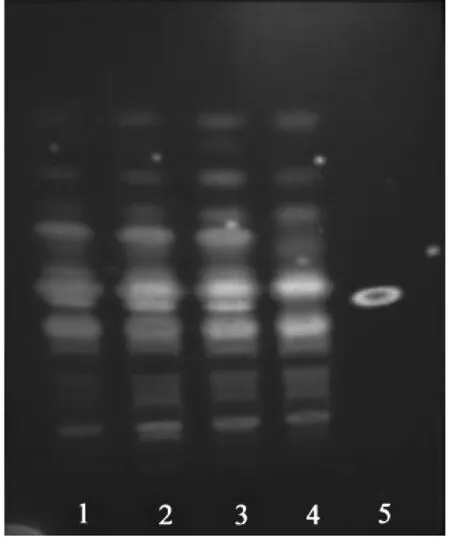
图1 延胡索TLC图 (紫外灯下观察)
2.1.2 白及的鉴别 取胃康散9 g,加70 %甲醇80 ml,超声处理30 min,滤过,滤液蒸干,残渣加水20 ml使溶解,用乙醚振摇提取2次,每次20 ml,合并乙醚液,挥至1 ml,作为供试品溶液[8]。取白及对照药材0.5 g及缺白及药材的阴性供试品9 g,同法制得白及对照药材溶液和缺白及的阴性供试品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验[7],吸取上述3种溶液各15 µl,依次点于同一硅胶G薄层板相应位置上,以环己烷-乙酸乙酯-甲醇(6:2.5:1)为展开剂,室温展开,取出,晾干后喷以10 %硫酸乙醇溶液,在105 ℃加热至斑点显色清晰,并且室温放置数小时。供试品色谱中,日光下检视在与对照药材色谱相应的位置上,显相同颜色的黄色斑点,阴性对照无干扰。见图2。

图2 白及TLC图
2.1.3 甘草的鉴别 取胃康散5 g,加乙醇20 ml,超声处理30 min,放冷,滤过,滤液蒸干,残渣加乙醇2 ml使溶解,作为供试品溶液[9-11]。取甘草对照药材2 g及缺甘草的阴性供试品5 g,同上法制得甘草对照药材溶液和缺甘草的阴性供试品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验[7]。吸取供试品溶液和缺甘草的阴性供试品溶液各10 μl、甘草对照药材溶液5 μl,分别依次点于同一硅胶G薄层板相应位置上,以三氯甲烷-甲醇-水(40:10:1)为展开剂(充分预饱和),室温展开,取出。晾干后喷以10 %硫酸乙醇溶液,并在105 ℃条件下加热至斑点清晰。供试品色谱中,日光下检视在与对照药材色谱相应的位置上,显相黄色的主斑点,阴性对照无干扰。见图3。
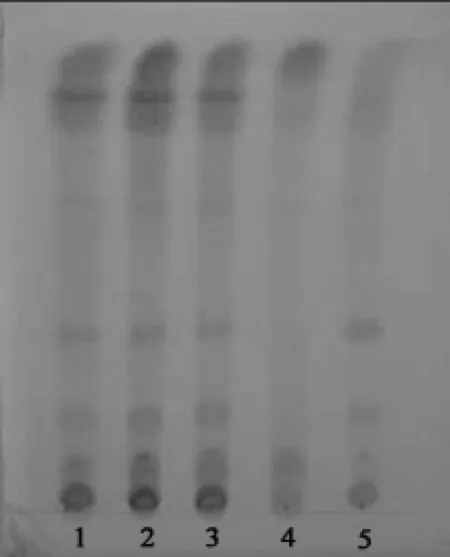
图3 甘草TLC图
2.2 氯化两面针碱的含量测定
2.2.1 色谱条件 Agilent 5 TC-C18柱(250 mm×4.6 mm,5 µm);流动相:甲醇-0.1 %磷酸(用N,N-二乙基乙胺调pH值6.0)(60:40);检测波长:273 nm;流速:1.0 ml/min;柱温:25 ℃;进样量10 µl[12]。
2.2.2 对照品溶液 取氯化两面针碱适量,精密称定,置200 ml量瓶,加80 %甲醇制成76.1 μg/ml的对照品储备溶液。用移液管精密吸取对照品储备液5 ml至20 ml量瓶,加80 %甲醇稀释至刻度,即得。
2.2.3 供试品溶液 取胃康散精密称约10.0 g,样品置100 ml锥形瓶,加入80 %甲醇30 ml,超声处理30 min,室温放冷,滤过至50 ml量瓶,并用80 %甲醇适量3次润洗锥形瓶和滤纸,并定溶至刻度,摇匀后用0.45 μm微孔滤膜滤过,取续滤液,即得。
2.2.4 阴性样品溶液 按处方量配制不含两面针的阴性样品,再按2.2.3项下方处理,即得。
2.2.5 专属性考察 分别取对照品、供试品和阴性样品溶液各10 μl,以2.2.1项下条件进样,结果见图4,氯化两面针碱约在6.3 min出峰,在该测定条件下,供试品中氯化两面针碱峰与杂质分离度良好,阴性样品中氯化两面针碱出峰位置无杂质峰的干扰,方法专属性好。
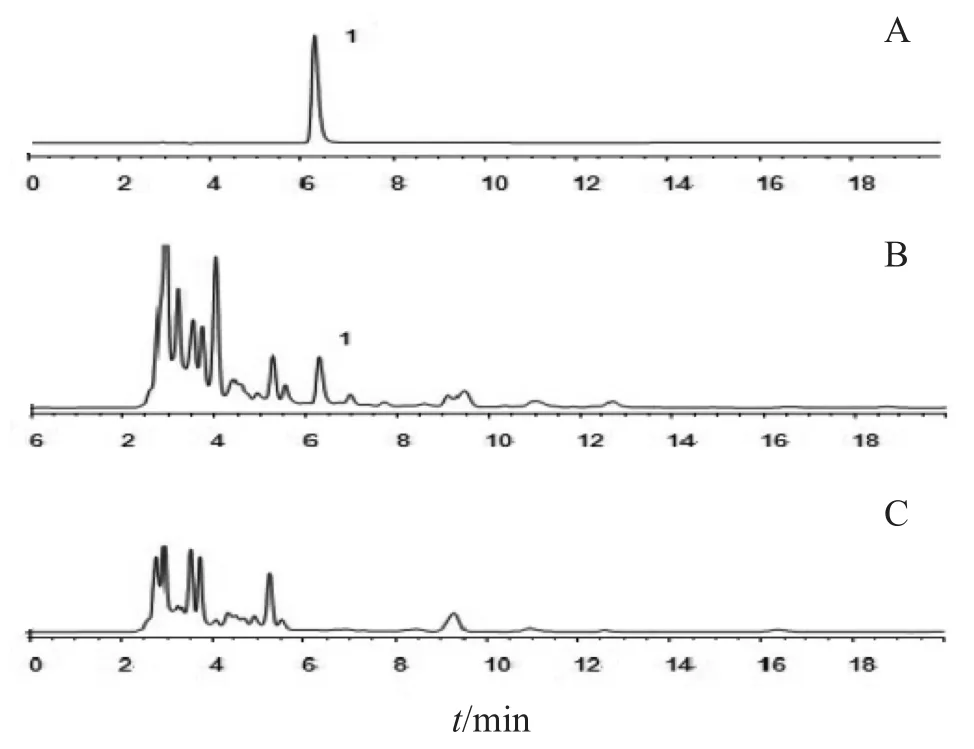
图4 胃康散专属性色谱图
2.2.6 线性关系考察 分别精密取贮备液1.0,2.0,4.0,6.0,8.0,10.0 ml,置入10 ml量瓶,加80 %甲醇定容至刻度,得浓度依次为7.61,15.22,30.44,45.66,60.88,76.1 μg/ml的对照品溶液[13]。按2.2.1项下条件,精密吸取上述系列溶液各10 μl顺次注入液相色谱仪。进行线性回归分析,以氯化两面针碱质量浓度(X)为横坐标,峰面积(Y)为纵坐标,得标准曲线为Y=54.67X-2.335,r=0.9995 (n=6),氯化两面针碱在7.6~76.1 μg/ml范围内线性关系良好。
2.2.7 精密度试验 按2.2.1项下色谱条件,精密吸取2.2.6项下浓度为45.66 μg/ml的氯化两面针碱对照品溶液10 µl注入液相色谱仪,连续进样6次,测定峰面积。其RSD为0.57 %,表明该系统及方法精密度良好。
2.2.8 稳定性试验 取胃康散(批号:171130)适量,按2.2.3项下的方法制备供试品溶液,分别放置于第0,2,4、6,8,10,12,14,16,18 h进样测定,记录色谱峰面积并计算含量,得氯化两面针碱峰面积RSD为0.93 %,表明样品溶液在室温放置条件下18 h内稳定。
2.2.9 重复性试验 取胃康散(批号:171130)6份,精密称定每份10.0 g,按2.2.3项下方法操作。再按2.2.1项下方法平行操作6次,该批号氯化两面针碱的平均含量为82.73 μg/g,RSD为2.1 %,表明该系统和方法的重复性良好。
2.2.10 加样回收率试验 精密称取胃康散(批号:171130)9份,每份约5.0 g,精密加入对照品贮备溶液适量,按2.2.3项下操作方法提取,再按2.2.1项下方法测定,计算平均加样回收率。结果见表1。

表1 氯化两面针碱加样回收率试验结果(n=9)
2.2.11 样品含量测定 精密称取胃康散3批(批号:171130、170331、180424)各适量,按2.2.3项下方法制备供试品溶液,然后按所建方法进样测定,计算样品中氯化两面针碱含量分别为0.2440,0.2972,0.2676 mg/袋。
3 讨论
3.1 薄层鉴别
延胡索薄层色谱鉴别中,笔者参考《中国药典》2015年版一部延胡索药材薄层鉴别项下方法,发现上述条件展开效果尚可,但主斑点的分离效果欠佳,背景太深。随后笔者比较了不同的展开剂系统正己烷-三氯甲烷-甲醇(7.5:3.5:1)、环己烷-乙醚-甲醇(3:5:0.5),结果以正己烷-三氯甲烷-甲醇(7.5:3.5:1)为展开剂,用碘蒸气熏至各斑点显色清晰,数小时(约6 h)挥尽板上吸附的碘后,在紫外灯(365 nm)下检视,其斑点显色清晰,且阴性样品无干扰。白及的鉴别采用了《中国药典》(2015年版)一部白及药材的薄层鉴别方法,本次未加修改,其色谱图中黄色斑点(比移值约0.385)为主斑点,阴性对照样品也无干扰。本次甘草鉴别,其样品采取超声的方式进行提取,最后喷以10%硫酸乙醇溶液,并在105 ℃加热至斑点清晰,立即置日光下检视,样品与甘草对照药材色谱中的2个黄色斑点(比移值约0.255、0.375)为主斑点,其阴性对照样品也无干扰。
3.2 HPLC含量测定溶剂提取的选择
根据氯化两面针碱[14-16]的化学性质,实验中分别考察了(20 %、50 %、80 %)甲醇、乙醇,也尝试了利用浓氨试液-甲醇(1:20)混合提取氯化两面针碱[17]。结果以80 %甲醇、乙醇超声处理30 min提取氯化两面针碱,测定峰面积较大。综合考虑,最终选定了80 %甲醇作为提取溶剂,其色谱图杂质峰较少,与相邻的色谱峰有很好的分离度。
3.3 HPLC含量测定中检测波长及流动相的选择
本研究的检测波长,直接选择了《中国药典》2015年版一部两面针药材含量测定项下的检测波长。流动相先后考察了乙腈-水、甲醇-水、乙腈-0.1 %磷酸水溶液、甲醇-0.1 %磷酸水溶液系统[18-21],试验过程中发现,氯化两面针碱峰型较宽,存在拖尾现象,只有选择甲醇-0.1 %磷酸溶液用作流动相,拖尾现象得以解决,分离效果好,但峰型较宽。随后笔者将0.1 %磷酸水溶液pH值用N,N-二乙基乙胺调为6.0时,其色谱峰与其他杂质峰分离度最好,保留时间适中,峰形好,故本试验流动相得以确定。
