中枢神经系统α7烟碱型乙酰神经胆碱受体显像剂研究进展
2019-06-14张华北
高 航,张华北
(放射性药物教育部重点实验室 北京师范大学 化学学院,北京 100875)
乙酰胆碱(acetylcholine, Ach)是广泛分布于生物体内的一种具有兴奋功能的神经递质。乙酰胆碱通过神经末梢中的乙酰胆碱转化酶催化乙酰辅酶A及胆碱进行生物合成。乙酰胆碱是神经元之间信息传递的关键物质。而乙酰胆碱受体(acetylcholine receptor, AChR)更是药物研发中重要靶点之一,乙酰胆碱受体已被发现与很多重大疾病及多种疾病所产生的病理生理现象有密切关系[1-2]。
影像学对阿尔兹海默症诊断方法主要分为两种:(1) 以传统CT、MRI为主的结构解剖影像学;(2) 以PET、SPECT、扩散张量显像(diffusion tensor imaging, DTI)、功能磁共振波谱显像(functional magnetic resonance spectroscopy, fMRI)等的功能影像学。目前应用于临床的PET显像剂主要是以18F-FDG为主的代谢显像剂,其优点为能明确脑能量代谢过程及途径,缺点为轻度AD患者难检测到,不适用于常规老年痴呆症治疗效果评价。
为了解决如上问题,乙酰胆碱受体显像剂的发现与发展为我们在诊断AD病人以及评价治疗效果上起到了重要作用。乙酰胆碱受体显像剂应有如下特点:高亲和力、良好的选择性、血脑屏障(blood brain barrier, BBB)透过能力、在脑内适宜的滞留等。本文总结了近几年来不同骨架的α7烟碱型乙酰胆碱受体显像剂的研究现状及进展。
1 乙酰胆碱受体的分类与结构
乙酰胆碱受体可按照在突触后膜产生不同生物学效应分为毒覃碱型(mAChR)和烟碱型乙酰胆碱受体(nAChR)。两种类型均广泛分布于中枢神经系统和外周神经系统[3]。其中毒覃碱型乙酰胆碱受体属于G蛋白偶联受体家族,并可以被覃毒碱激动,被阿托品特异性阻断。而烟碱型乙酰神经胆碱受体,属于半胱氨酸环配体门控的离子通道受体家族,可以被烟碱(nicotine)激动,而被箭毒阻断。目前已经克隆出的nAChR蛋白单体共有十余种,包括在中枢神经系统的α2~α10及β2~β4;以及在外周肌肉型的α1,β1,δ,γ,ε型单体。α7型nAChR是由5个相同的α7亚基构成的五聚体跨膜蛋白。α7 nAChR对钙离子有较高的通过性,可以调节钙离子在细胞膜膜内外的浓度,以及乙酰胆碱的释放。研究表明,α7 nAChR与多种中枢神经系统疾病密切相关,例如老年痴呆症(alzheimer disease, AD)、癫痫(epilepsy)、帕金森病(parkinson’s disease, PD)、精神分裂症(schizophrenia)、炎症(inflammation)、动脉粥样硬化(atherosclerosis, AS)等密切相关[4-9]。
2 α7烟碱型乙酰胆碱受体与退行性神经疾病
研究表明AD、PD等退行性神经疾病与α7 nAChR有密切关系。用免疫组化、放射性配体受体结合实验等方法对α7 nAChR在体内密度研究表明,在退行性神经疾病中发现了α7 nAChR的减少[10]。
此外,AD的主要病理特征之一为,在大脑海马区和大脑皮层出现β样淀粉蛋白沉淀聚集而形成的老年斑,随着研究的深入,更多证据表明Aβ1-42在脑中会部分与神经性的α7 nAChRs结合[11-12],这种结合促使脑中聚集Aβ1-42-α7 nAChRs复合物[13],并迅速使Tau 蛋白磷酸化[14],从而使α7 nAChRs离子通道受到严重阻滞或破坏,最终导致胆碱能神经递质损伤[11]以及神经细胞死亡[15],临床表现为影响记忆和认知障碍[16]。这些研究都表明在AD病人脑中的α7 nAChRs被Aβ42慢性破坏从而导致神经缺陷和障碍,最终造成Aβ斑块和磷酸化的神经纤维缠结(phosphorylated nerve fiber tangles, NFTs)沉淀聚集。
Lee等[17]研究发现,除了淀粉样蛋白前体(APP)的大量存在和淀粉样的沉淀聚集,缺失α7 烟碱型乙酰神经胆碱受体还会导致AD模型老鼠中突触的完整性(通道和生长),进而影响学习和记忆等活动行为[18]。因此,干扰Aβ1-42-α7 nAChRs的相互作用可以减少Aβ1-42调节中的功能缺陷,神经退行行为以及其他可能的AD临床病理表现。卢家红等[19]采用免疫组化单染方法和共染方法观察了α7 nAChRs在AD患者脑中沉积的情况与Aβ1-42的关系,研究结果表明,在AD患者脑中有α7 nAChRs的异常沉积,并且分布与Aβ1-42形成的老年斑的部位大致相同,后续实验发现了α7 nAChRs与Aβ1-42的特异性结合可以被α7 nAChRs的激动剂所拮抗。这些结果都表明了α7 nAChRs在Aβ1-42调节的病理生理学方面都起到了非常重要的作用。而α7 nAChRs的激动剂也将成为潜在的治疗以AD为主的退行性神经疾病药物。
在基因剔除、亚型选择性配体等方面的研究表明,靶向性α7 nAChRs受体激动剂能够提高认知能力以及改善听觉门控缺陷。PNU-282987[20-21]、PHA-543613[22]、ARR17779,SSR180711[23]以及A-582941[24]等高选择性的α7 nAChRs受体激动剂提高了感觉-门控缺陷、改善了短期工作记忆、以及记忆固化等模型的认知功能。
3 α7烟碱型乙酰胆碱受体配体及激动剂研究现状
在近年来研究中,高选择性的α7烟碱型乙酰神经胆碱配体结构不断发展,开拓出了不同骨架的结构。最初的结构主要集中于假木贼碱衍生物(GTS-21[25])和螺环恶唑烷酮类衍生物(AR-R17779[26])。近年来文献中又报道了若干新骨架,如以氮杂双环二胺为骨架的结构以及奎宁环氨基甲酸酯[27]、奎宁环酰胺[28](PHA-613,543和PHA-487,568[22,29])、奎宁环醚[30]以及1,4-二氮杂环[3,2,2]壬烷取代的衍生物(SSR180711[23,31-32]),以及A-941,582[33]、PHA-829,709[34](如图1)。
在所发现的高亲和力配体中,部分α7 nAChRs的激动剂已经作为退行性神经疾病治疗药物进入临床试验,例如GTS-21(DMXBA)。DMXBA是天然产物假木贼碱的衍生物,并于2003年开始了Ⅰ期临床试验;在2006年底进入了Ⅱ期临床试验[34]。另一个选择性α7受体激动剂SSR180711目前也已经进入了临床试验[35]。SSR80711在老鼠和人类α7受体亲和实验中都表现出了高亲和性(Ki=(22±4);(14±1) nM)。除此之外,其余一些选择性的α7 nAChRs 药物如PHA-543613、AR-R17779、A-582941等都已经进行了动物和人体实验,实验表明其都能有效提高感觉-门控缺陷、改善短期工作记忆、以及改善记忆固化等认知功能。因此,这些配体化合物有望成为潜在的α7 nAChRs靶向性的中枢神经系统疾病治疗药物以及放射性配体的先导化合物。
为了实现AD的早期诊断,用于活体诊断放射性核素显像剂的研发至关重要。体内显像核素主要有PET显像的正电子核素11C、18F、76Br,以及用于SPECT显像的单光子核素123I、125I等,将放射性核素标记相应配体后获得的高选择性、高亲和力的靶向α7烟碱型乙酰胆碱受体的放射性配体化合物可用于以AD及PD为例的神经退行性疾病的诊断。
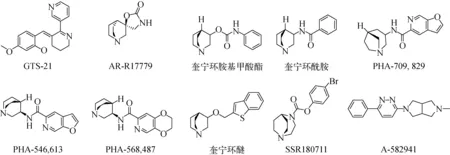
图1 α7烟碱型乙酰胆碱受体配体激动剂结构Fig.1 Chemical structure of α7nicotinic acetylcholine receptor agonists
4 α7烟碱型乙酰胆碱受体放射性配体研究进展
4.1 喹宁环及喹宁环酰胺为主的α7 nAChR显像剂
最早报道的α7nAchR放射性配体为两种碘标记物以及一种氚标记化合物。[125I]α-bgt[36](又名银环蛇毒素,Kd=(1.5±0.7) nM)。[125I]MLA[37](又名甲基牛扁碱,Kd=(1.8±0.4) nM) 以及[3H]MLA[38](甲基牛扁碱,Kd=(1.86±0.31) nM),它们与α7烟碱型乙酰胆碱受体均有较高的特异性结合,而且在脑中的浓度分布也基本相似。但是,α-bgt和MLA分别来源于天然物质抗银环蛇毒血清和百合花种子,均具有较大毒性。且两种物质均为生物大分子,难以透过血脑屏障,不能用于正常的体内研究。且不具有标记18F等正电子核素的位点,故不能发展成为合适的α7 烟碱型乙酰神经胆碱受体体内显像剂。但鉴于其高选择性和亲和力,目前主要用于进行体外活性测定等研究。
近几年当中,α7 nAChR的放射性配体获得了不断的发展,尤其是正电子核素标记的放射性配基。
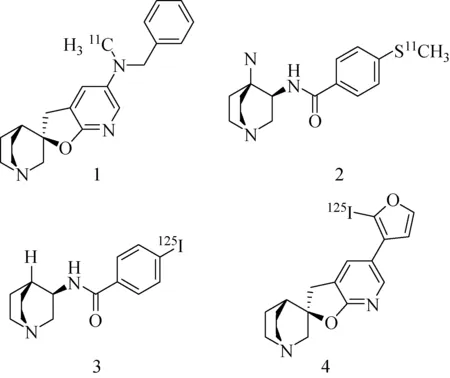
图2 四种放射性配体[39]Fig.2 Radioactive ligands[39]
2005年,Pomper等[39]报道了四种放射性配体(图2),4个化合物Ki抑制常数分别为0.54、5.8、18、0.26 nM。化合物1、2、3在脑中的吸收值都较低并且没有区域选择性。而化合物4能够在有效进脑的情况下还获得一定的靶向性吸收,但其BPND值(靶向性区域吸收/小脑吸收-1)仅为0.6,并不适合作为进一步的显像剂进行研究。
2006年,Ogawa等[40]研究了单光子放射性显像剂[125I]I-TSA(图3),其取代物I-TSA和Br-TSA的亲和力Ki值分别为0.54 nM和1.11 nM,有较高的特异性和选择性。同时,[125I]I-TSA在脑区域分布也符合α7 nAChRs在脑中的分布,在海马中较高,小脑中最低,且小脑中的清除速率也明显快于其他的靶组织。但是,在药物自阻断实验当中,非标记的I-TSA和MLA均不能够抑制放射性吸收。虽然[125I]I-TSA拥有较高的受体亲和力、选择性、脑摄取以及代谢稳定性等性质,但是它较高的非特异性结合也证明了它不适合进一步作为α7 烟碱型乙酰胆碱受体放射性显像剂。
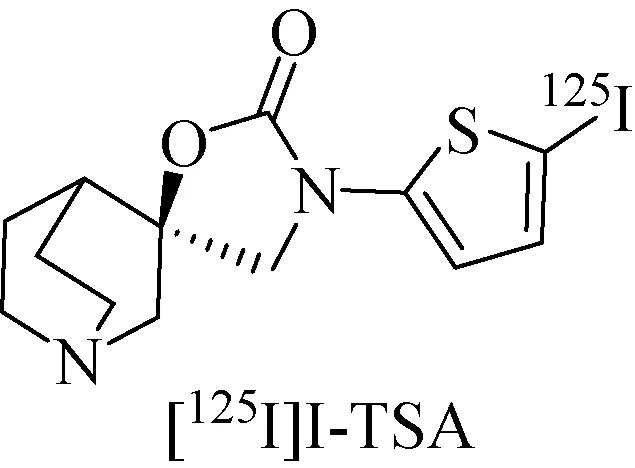
图3 [125I]I-TSA[40]Fig.3 [125I]I-TSA[40]
4.2 α7 nAChR激动剂标记
GTS-21作为α7 nAChRs的部分激动剂,能够有效提高以AD为主的退行性神经疾病患者的认知能力。而4-OH-GTS-21是GTS-21在体内的主要代谢产物之一,该化合物也被证实有较好的药效,据此Kim等[41]在2007年对GTS-21的不同取代基位置以及它的两个体内代谢产物进行了11C标记,得到了四种正电子标记的化合物物(图4),同时研究了它们的动力学性质和体内PET显像。研究结果表明,在狒狒脑中,[2-甲氧基-11C]GTS-21和[4-11C]GTS-21有高且快的初始摄取(1~3 min)但是体内清除很快,滞留性较差;然而30 min以后由于[4-11C]GTS-21的体内代谢产物进入脑中,清除减缓。[2-甲氧基-11C]4-OH-GTS-21、[2-甲氧基-11C]2-OH-GTS-21表现出了相对比较好的脑摄取与滞留,并且相对于4-羟基取代位置,2-羟基取代位置更适合作为热标记取代位进行药代动力学研究,且该化合物更容易透过血脑屏障。
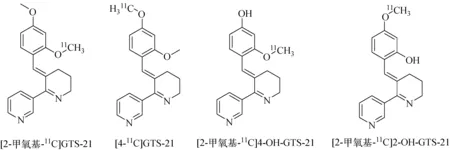
图4 11C标记的GTS-21衍生物[41]Fig.4 11C-labeled GTS-21 derivatives[41]
4.3 1,5-二氮杂二环[3.2.2]壬烷及六氢吡咯并[3,4]吡咯为母核的柔性α7 nAChR显像剂
除GTS-21外,另外一种α7 烟碱型乙酰胆碱受体部分激动剂SSR180711也获得了进一步的研究,日本千叶大学设计并合成标记了一系列正电子及单光子SSR180711放射性衍生物,分别为[11C]CHIBA-1001、[76Br]SSR180711、[125I]CHIBA-1001、[3H]CHIBA-1001、[131I]CHIBA-1001,在其中对[11C]CHIBA-1001的研究最为深入,亦为首个进入到人体试验的α7 nAChRs放射性显像剂。
2008年,Kenji[42]首次报道了[11C]CHIBA-1001和[76Br]SSR180711(图5)的研究成果。对于[76Br]CHIBA-1001,PET显像实验表明了它能够快速透过血脑屏障,迅速聚集在颅内并于60 min时在海马摄取达到峰值。同时该化合物在脑中各区域的分布也与α7 nAChRs相一致,阻断实验也表明它是一个高选择性的α7 nAChRs示踪剂。

图5 示踪剂[76Br]SSR180711与[11C]CHIBA-1001化学结构[42]Fig.5 Chemical structure of [76Br]SSR180711 and [11C]CHIBA-1001[42]
另外一种正电子示踪剂[11C]CHIBA-1001则显示出了更为优良的性质,30 min时,其在非人类灵长类动物的海马中的吸收达到了峰值。但综合考虑核素的生产、核素自身性质(76Br半衰期:16.2 h远大于11C:20.41 min) 以脑内分布,[11C]CHIBA-1001的性质要优于[76Br]SSR180711。因此,[11C]CHIBA-1001可作为潜在的候选α7 nAChRs示踪剂。
2009年,Toyohara等[43]分别深入研究了[11C]CHIBA-1001在小鼠体内的生理分布、毒性、代谢性质等临床前研究内容,以及在人脑中的PET显像研究。人脑PET显像实验表明,[11C]CHIBA-1001迅速聚集在脑中各区域,且大致分布与α7 nAChRs相同,它的聚集度已经完全满足临床需求。相比于动物中的实验,[11C]CHIBA-1001在人体血液中也有良好的稳定性,在注射60 min后仍然有超过80%的放射性以自身形式存在。这些实验表明[11C]CHIBA-1001适合更进一步进行人脑显像的临床研究。
在2011年,日本千叶大学又进一步对[11C]CHIBA-1001进行了人体实验。对三位22~24岁无烟史、无神经退行性疾病的女性志愿者进行了研究。[11C]CHIBA-1001在人体中的分布,与动物分布中的数据略有差异。它主要通过肝胆系统和肠胃系统来进行代谢,因此在人体小肠中的吸收最高。而在啮齿类动物实验中,最高吸收值出现膀胱,说明为肾脏代谢。人体代谢实验中,通过检测志愿者的尿液,发现大部分的放射性仍以自身的形式存在,而在血清中存在92%(鼠中只有21%)。研究人员通过计算发现,[11C]CHIBA-1001在人体中的有效剂量为6.9 μSv/MBq,其结果符合辐射安全的范围而能够进行下一步的临床研究[43]。
同年,日本千叶大学的Masatomo Ishikawa等[44]进行了人脑阻断研究。研究了两种药物托烷司琼(20 mg)和昂丹司琼(8 mg)对人脑显像的阻断作用。结果表明,20 mg Tropisetron虽对[11C]CHIBA-1001在人脑中的吸收有抑制作用,但并不完全。同时,当注射[11C]CHIBA-1001后,虽然其迅速充满整个大脑,但缺乏一个横断面来确定BPND值,更重要的是,采用体外人脑样本的匀浆进行受体结合实验时发现CHIBA-1001的非特异性结合非常高,因此该药物有待继续研究。
2010年,Toyohara等[45]报道了两个正电子核素11C标记的α7 nAChRs显像剂[11C]A-582941和[11C]A-844606(图6),其抑制常数Ki分别为10.8 nM和11 nM。在啮齿类动物体内分布实验中,前者表现出了较高的非特异性结合,但在清醒的猴子脑中,[11C]A-582941和[11C]A-844606可以被预注射的5 mg/kg SSR180711特异性抑制。因此[11C]A-582941和[11C]A-844606也可以作为潜在的α7 nAChRs显像剂。
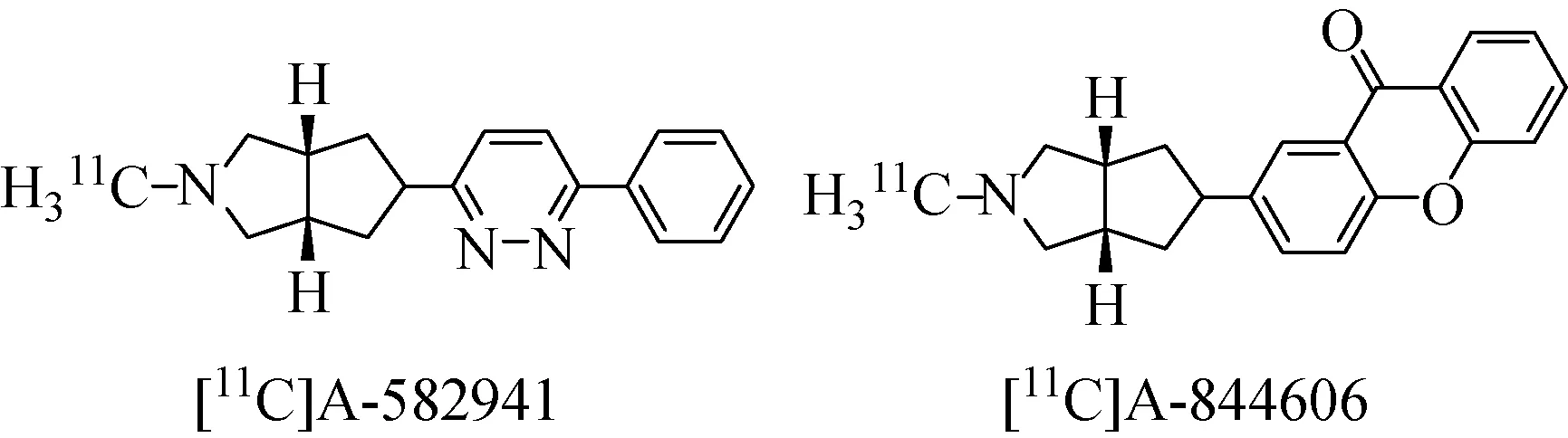
图6 显像剂[11C]A-582941和[11C]A-844606化学结构[45]Fig.6 Chemical structure of [11C]A-582941 and [11C]A-844606[45]
2010年Ogawa等[46]又设计并合成了两种C-11标记的α7 nAChRs放射性配体:[11C](R)-MeQAA(Ki=(73.7±3.5) nM)和其对应异构体[11C](S)-MeQAA(Ki=(186.3±25.7) nM)(图7)。它们对α7 nAChRs的亲和力较差,不如[11C]CHIBA-1001(Ki=49 nM), 且对5-HT3受体的选择性也较差。
生物分布实验表明,二者的初始脑吸收都较高,且R构型略高于S构型。在接下来的猴脑PET显像实验中,两种化合物都快速聚集在丘脑和皮层等区域,但绝对值都不高,当用SSR180711阻断时获得了一定的抑制效果,但5-HT3受体配体Ondansetro也获得了同样的效果。因此,它们并不适合作为α7 nAChRs PET显像剂,需要进一步优化结构。

图7 显像剂[11C](R)-MeQAA和[11C](S)-MeQAA化学结构[46]Fig.7 Chemical structure of [11C](R)-MeQAA and [11C](S)-MeQAA[46]
2011年, Ettrup等[47]将α7 nAChRs的高亲和力的选择性激动剂NS14492(Ki=2.2 nM)进行了C-11标记得到了[11C]NS14492(图 8),并进行了猪脑PET显像。[11C]NS14492在猪脑的皮层、海马、丘脑等区域聚集最多,而在小脑最少,预注射两种不同结构的α7 nAChRs阻断剂SSR180711和NS14492时,在脑内所有区域的VT值均减少。因此,[11C]NS14492也可以作为一个潜在的α7 nAChRs显像剂。
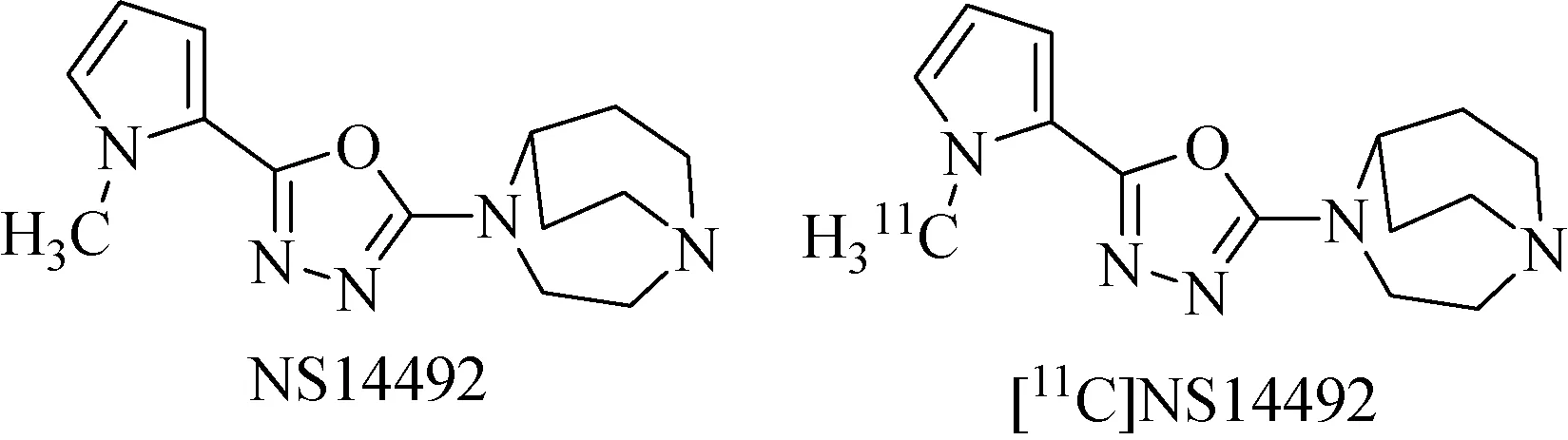
图8 示踪剂[11C]NS14492及其非放化合物NS14492化学结构[47]Fig.8 Chemical structure of [11C]NS14492 and NS14492[47]
同年,Briggs等[48]设计并合成了[18F]NS10743(图9)并通过PET显像研究了其在猪脑中各区域的分布情况。实验结果表明,[18F]NS10743能够快速透过血脑屏障(BBB)进入脑中,当用α7 nAChRs的部分激动剂NS6740[47]阻断时,SUVmax值变为105%~110%后迅速降低,说明了[18F]NS10743是以α7 nAChRs为靶向性的。
2013年,Deuther-Conrad等[49]又研究了与[11C]NS14492和[18F]NS10743相近的高亲和性化合物[18F]NS14490 (图10) 的基本理化性质,2014年又在其基础上进行了PET显像实验。当分别用α7 nAChRs高亲和性配体NS6740(10 mg/kg)和SSR180711(10 mg/kg)阻断该药物在脑中的吸收时,发现了NS6740减少了[18F]NS14490在脑中22%的吸收,而SSR180711却没有明显变化。
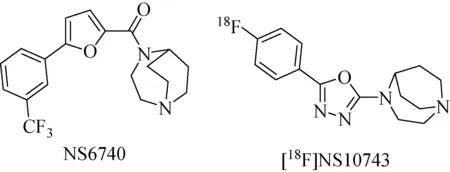
图9 示踪剂[18F]NS10743及α7 nAChRs高亲和性配体 NS6740化学结构[48]Fig.9 Chemical structure of [18F]NS10743 and NS6740[48]
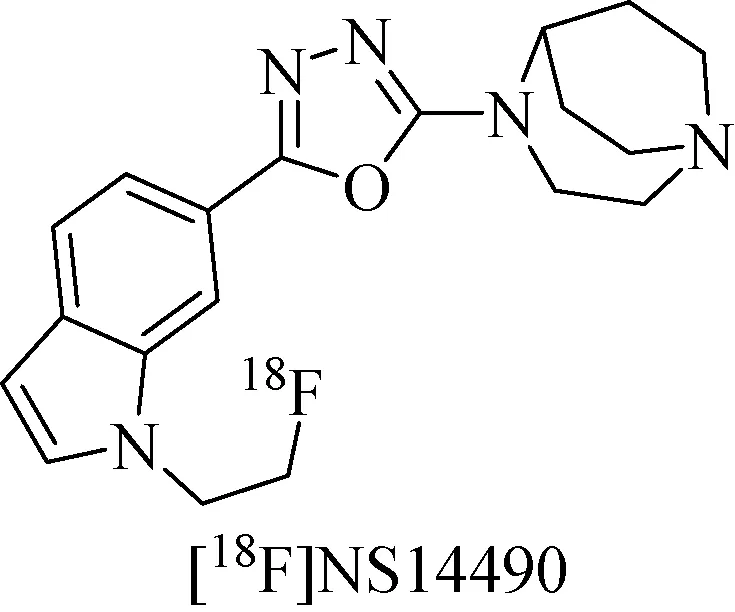
图10 示踪剂[18F]NS14490化学结构[49]Fig.10 Chemical structure of [18F]NS14490[49]
基于该化合物高的亲和性,2014年该实验组做了猪的PET显像实验[50]。通过PET显像,观察[18F]NS14490在脑中的吸收及分布。
综上,[18F]NS14490有一定希望成为潜在的α7 nAChRs显像剂。
2012年, Gao等[51]研究了([11C]rac-(1))(图 11)在鼠脑内的分布情况。实验表明,[11C]rac-(1)主要集中在额叶皮质、丘脑、纹状体和海马中。当用选择性α7 nAChRs激动剂PHA543616抑制时,可发现在小脑中的抑制作用是最佳的,在海马、额叶皮质和丘脑中都显示出了特异性的减少,这说明[11C]rac-(1)能够与α7 nAChRs特异性结合,可以成为一个潜在的α7 nAChRs PET显像剂。

图11 示踪剂[11C]rac-(1)化学结构[51]Fig.11 Chemical structure of [11C]rac-(1)[51]
2013年, Horti等[52]又发现了新的体外高亲和性α7 nAChRs示踪剂,[11C]A-833834(Ki=1.53 nM)和[11C]A-752274(Ki=0.092 nM)(图12)。实验表明[11C]A-752274在体外的受体亲和实验中体现出了非常高的亲和性。但它在接下来的小鼠体内分布实验中,绝对进脑量却较少,其绝对吸收值仍较低,结果并不理想。该实验组认为是亲水性太高(logD7.4=-2.7)导致药物无法透过血脑屏障,造成低进脑量,因此需要对该化合物进行结构上的改进。
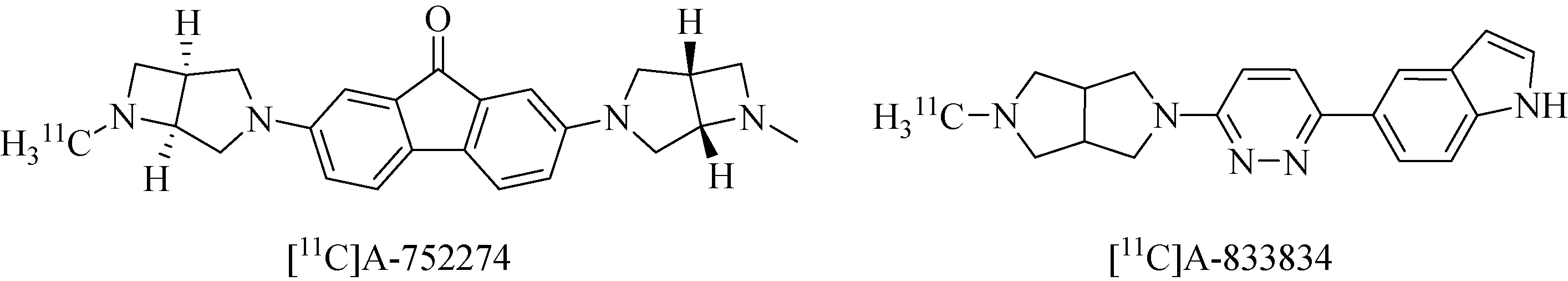
图12 示踪剂[11C]A-752274 和[11C]A-833834化学结构[52]Fig.12 Chemical structure of [11C]A-752274 and [11C]A-833834[52]
同年,Ravert[53]又报道了高亲和性α7 nAChRs配体[18F]AZ11637326(Kd=0.2 nM)(图13) 的研究内容。在生物分布实验中,[18F]AZ11637326可以迅速布满整个大脑,30 min时在海马的吸收达到最高,小脑中最低。当用MLA、烟碱和Ondansetro抑制时,却出现了吸收值的少量增加。实验结果说明,该化合物在鼠脑中有一定的特异性结合。但应用于狒狒脑显像时,基本没有发现特异性结合,因此该化合物并不能成为α7 nAChRs PET显像剂,还需要对其进一步优化。
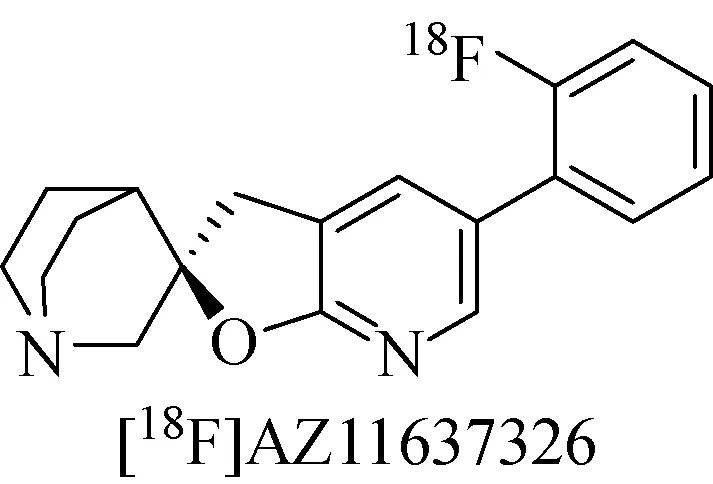
图13 示踪剂[18F]AZ11637326化学结构[53]Fig.13 Chemical structure of [18F]AZ11637326[53]
4.4 1,5-二氮杂二环[3.2.2]壬烷为母核取代的刚性二苯基二氧化噻吩及9-芴酮作为α7 nAChR显像剂
2013年, Gao等[54-55]发表了一系列高亲和性的二苯并噻吩类化合物 (Ki=0.4~20 nM),并对其中活性数据最好的两个化合物进行了放射性标记(图14)。这两种化合物对α4β2nAChRs和5-HT3也有很高的选择性,Ki,α7/α4β2=1 370,Ki,α7/5-HT3=561。在接下来的生物分布实验中,[18F]7a和[18F]7c都表现出了很高的初始摄取。
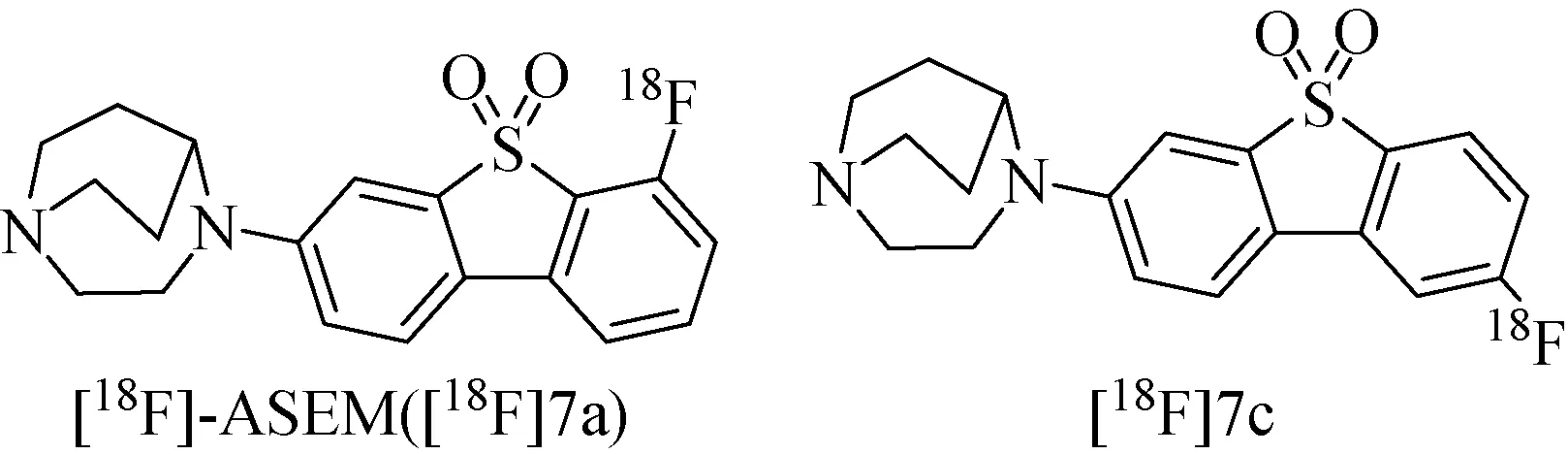
图14 示踪剂[18F]-ASEM 和[18F]7c化学结构[54-55]Fig.14 Chemical structure of [18F]-ASEM and [18F]7c[54-55]
基于[18F]7a的优良性质,该小组在2014年将其正式命名为[18F]-ASEM,并研究了它在DISC1老鼠和狒狒脑中的活体显像性质。实验结果表明,[18F]-ASEM能迅速进入狒狒的脑中,并与所有α7 nAChRs存在区域特异性结合 (特异性结合比例达到80%~90%)。高选择性的α7 nAChRs配体SSR180711对[18F]-ASEM 的吸收也起到了剂量依赖的抑制作用,这表明[18F]-ASEM在脑中的结合是与α7 nAChRs相关的,并适合做下一步的药物评价实验。狒狒脑中的显像实验也充分说明了[18F]-ASEM在各区域的高吸收,同时在一种精神分裂模型鼠脑内的结合则比正常鼠中的结合值要低很多,这也与之前报道的人类的情况相一致。因此[18F]-ASEM有希望成为量化人脑内α7 nAChRs的PET示踪剂。
在2017年,北京师范大学放射性药物教育部重点实验室张华北[56]课题组报道了高亲和性单光子125I标记的α7烟碱型乙酰神经胆碱配体[125I]CAIPE和[125I]IBT及[125I]IPPU(图15)。热标记实验表明,三种配体化合物均有较高的标记率(标记率>92.5%)。脂水分配系数数据表明具有良好BBB透过性(logP=1.2~1.7)。三种配体的体内外稳定性均良好。
[125I]IPPU体内生物分布实验显示出了很强的初始吸收,5 min时在脑中的吸收值达到(7.71±0.47)%ID/g,但是该化合物在小鼠体内的滞留性相对较差。[125I]IBT生物分布结果表现出了很强的初始摄取,在15 min时脑中的放射性吸收值为(8.36±0.57)%ID/g,并达到了最高;到30 min时,该吸收值仍然高于7%ID/g((7.32±0.59%)ID/g)。将[125I]IBT与已在人体做实验的药物[11C]CHIBA-1001相比,它也体现出了相对优越的吸收特性。因此放射性配体[125I]IBT 有希望成为潜在的α7 nAChRs 放射性配体。
2018年,北京师范大学放射性药物教育部重点实验室张华北等[57]又报道了对α7烟碱型乙酰神经胆碱受体高亲和性芴酮类衍生物配体YJF,YJF,YLN,YLF及其18F标记的放射性配体[18F]YLF(如图16,文中5,6,14,15)。四种配体都表现出了极高的受体亲和力,其中YLN亲和力高达0.0069 nM,为现有配体中活性最高的配体。放射性配体[18F]YLF具有合适的放射性化学特性,并具有较强的ɑ7 nAChRs亲和力(Ki=(2.98±1.41) nM),适合进行深入研究。
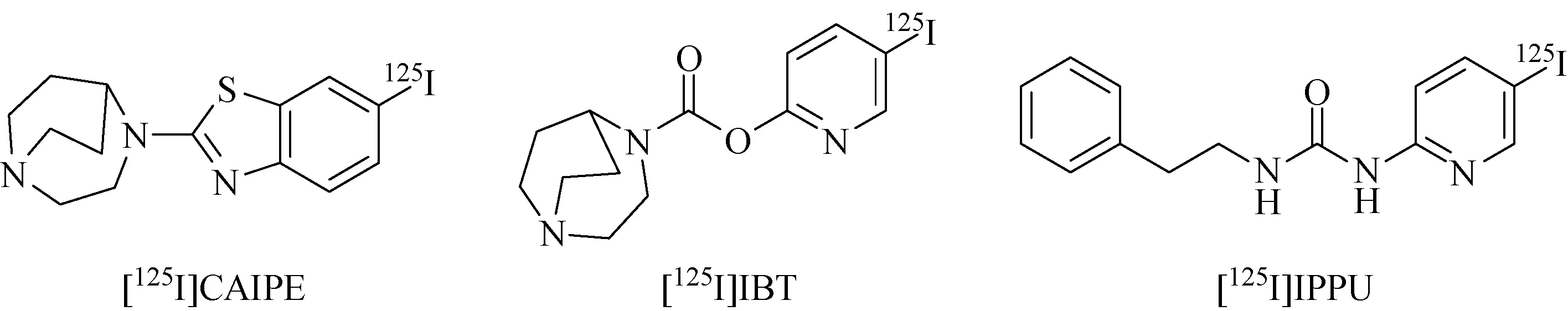
图15 示踪剂[125I]CAIPE、[125I]IBT及[125I]IPPU化学结构[56]Fig.15 Chemical structure of [125I]CAIPE、[125I]IBT and [125I]IPPU[56]
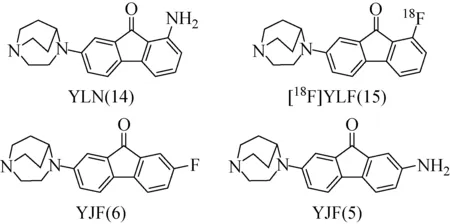
图16 配体YLN、YJF、YFN及示踪剂[18F]YLF的化学结构[57]Fig.16 Chemical structure of YLN、YJF、YFN and [18F][57]
在对[18F]YLF标记后进行体外稳定性实验,[18F]YLF在生理盐水和胎牛血清中都表现出很好的稳定性。生物分布实验说明,18F标记的放射性配体[18F]YLF在小鼠脑内具有非常高的初始脑摄取,在注射5 min后其摄取值即达到(8.98±0.41)%ID/g,15 min后显示出最高的脑摄取值(11.60±0.14)%ID/g;同时,该放射性配体表现出适宜的脑清除速率,在给药60 min、90 min后,其脑内的摄取值分别降为(5.46±0.27)%ID/g和(3.63±0.25)%ID/g,这表明了该化合物具有适宜的脑内动力学性质;该结果与目前已报道的进入临床实验的[18F]ASEM(其最高的脑摄取值出现在给药5 min后,为7.5%ID/g)相比,已表现出明显的优势。另外,[18F]YLF在血液中的摄取值很低,表现出很高的脑/血比值,在5 min和15 min时分别为9.35和9.57。脑区分布实验结果表明,该放射性配体在α7 nAChR最为富集的皮层、纹状体和海马区有较高的吸收,并在给药30 min后达到峰值。该区域分布特点与文献中报道的α7 nAChR在体内外的分布情况一致。脑内抑制实验证明,[18F]YLF对于α4β2nAChR和5-羟色胺受体几乎没有结合,该放射性配体对于α7 nAChR具有良好的选择性。
随后研究成员进行了大鼠PET显像研究(图17),显像结果表明,该放射性配体具有较高浓度的脑摄取,并具有适宜的脑部滞留,适于进行PET显像研究。另外,研究成员还对YLF标准品进行了急毒实验,结果表明得其半致死剂量LD50为53.70 mg/kg,临床上做一次PET显像所注射的剂量数量级别远小于LD50值使用安全。这些优良的性质均表明[18F]YLF可以作为潜在的α7 nAChR PET显像剂进行深入研究。
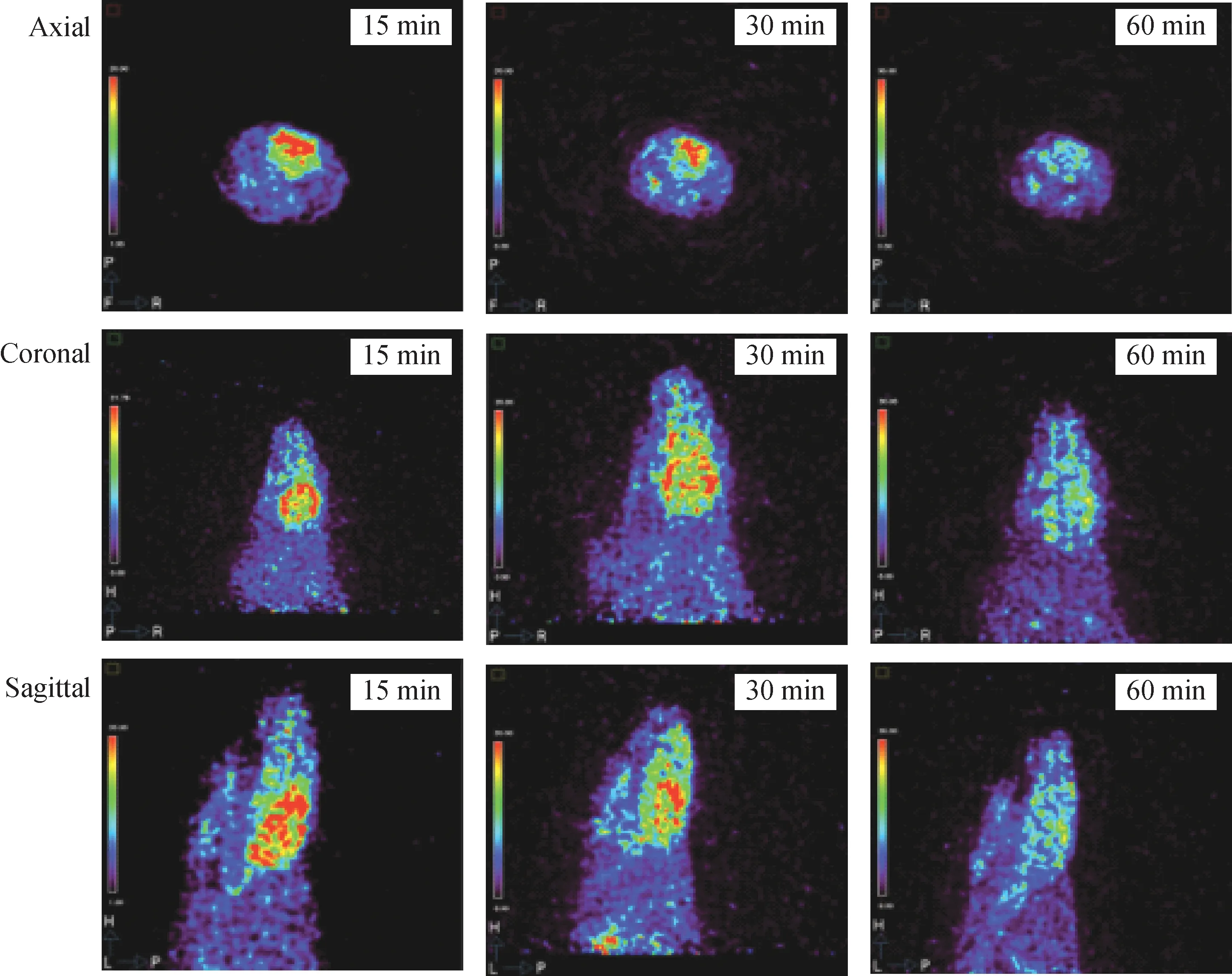
图17 [18F]YLF雌性CD-1大鼠PET显像图(红色区域为α7 nAChR)Fig.17 PET imaging of [18F]YLF in female CD-1 Rat(α7 nAChRis shown in red)
5 总结
综上所述,在几类用于α7 nAChRs的显像剂中,以1,5-二氮杂二环[3.2.2]壬烷为母核取代的刚性二苯基二氧化噻吩及9-芴酮作为α7 nAChRs的显像剂性质最好(如[18F]-ASEM、[18F]-YLF)。其优点主要表现为对靶点的高亲和力、高选择性及进脑速度快、滞留好等体内动力学性质。而对于1,5-二氮杂二环[3.2.2]壬烷为母核取代的柔性结构(如[11C]-CHIBA-1001),虽然容易通过血脑屏障,但都或多或少存在选择性差、特异性低等问题。而对于喹宁环及喹宁环酰胺取代化合物,不良的血脑屏障透过性及较高非特异性结合是阻碍其发展的主要原因。
PET/SPECT技术的发展为老年痴呆的早期诊断和治疗提供了新的机遇,放射性核素种类多样,各有所长。目前尚未有完成用于AD早期检测临床试验的药物,但结合多种放射性配体检查可辅助临床的鉴别诊断。近年来对α7nAChRs放射性配体的深入研究都为更好地理解退行性神经系统疾病的病理生理以及疾病的进程提供了更先进的研究手段。我们期望在未来有更多更深入的研究,为AD早期诊断和病程检测提供新思路。
