4-碘-2,3,5,6-四氟-甲基苯与3-甲基吡啶、呋喃及丁酮间卤键作用的理论研究
2019-06-03周芳芳王金树张玉平王清河单秀华
周芳芳,王金树,张玉平,王清河,单秀华
(1.承德石油高等专科学校 a. 石油工程系;b. 工业中心,河北 承德 067000; 2.吉林大学 化学学院,吉林 长春 130012)
与氢原子类似,卤素原子(Cl、Br、I)也可作为电子接受体与电子给体(N、O、S等)发生非共价相互作用,形成卤键[1]。卤键在分子识别与组装、药物设计、晶体工程、材料科学等领域均有重要应用[2-5]。在有机合成中,吡啶、丙酮呋喃及它们的衍生物通常作为非常重要的工业原料而被广泛应用;在分子层层组装中,更是作为卤键的重要的电子给体[6]。本文选取了PIP作为电子接受体,MPY、MEK和THF作为电子给体,对其形成的卤键复合物进行了理论研究。通过运用高水平的量子化学计算方法,讨论了复合物的结构特征、相互作用能、电荷密度拓扑性质和自然键轨道作用特征,从而揭示卤键相互作用的内在结构特征和相互作用的本质。
1 计算方法
为找到适合描述卤键体系的泛函,文中采用密度泛函方法X3LPY、B3LPY方法和从头算MP2方法对复合物结构进行优化并在相同水平下计算其频率,得到复合物的稳定构型。计算结果表明,B3LYP方法更加适合描述本复合物体系,因此在后续计算中,都采用B3LPY泛函。计算中使用了6-311++G(d, p)-PP基组,其中的赝势基组PP用来描述I原子的内核轨道,6-311++G(d, p)用来描述除I原子之外的所有原子的轨道组成[7]。相互作用能(ΔE)定义为复合物优化后的能量与相互作用两部分的优化结构能量之差。考虑到基组重叠误差(BSSE)对MP2方法造成的影响采用CP方法进行能量的校正[8]。以上计算均通过Gaussian09程序包完成[9]。电荷密度拓扑性质分析通过AIM2000程序包完成;自然键轨道作用分析包含于Gaussian09中的NBO3.0程序完成。
2 结果与讨论
2.1 计算方法的选择
采用X3LYP、B3LPY和MP2方法优化得到稳定结构,其结构相似;得到的二聚体的相互作用能列于表1中。
从表1可以看出,与高精度的MP2方法相比,X3LYP方法明显高估了复合物的相互作用能,因此不适合用来计算卤键体系的相互作用能;B3LYP泛函得到的结果与MP2结果非常接近,说明B3LYP泛函更适合于描述卤键作用的结构。因此后面均采用B3LYP泛函进行计算。
表1 不同方法下复合物的相互作用能kcal·mol-1
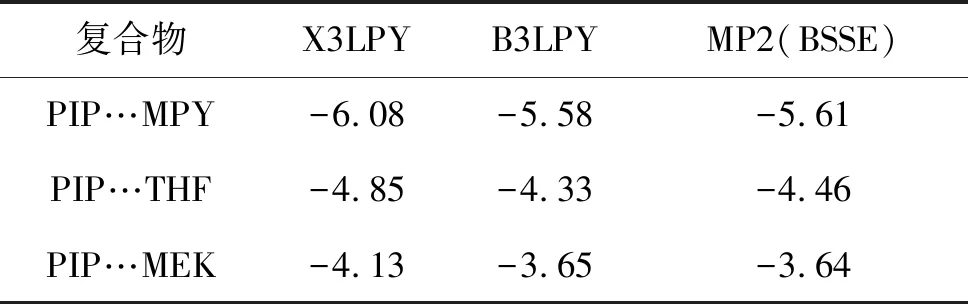
复合物X3LPYB3LPYMP2(BSSE)PIP…MPY-6.08-5.58-5.61PIP…THF-4.85-4.33-4.46PIP…MEK-4.13-3.65-3.64
2.2 结构与能量分析
经B3LPY优化的PIP…MPY、PIP…THF和PIP…MEK复合物平衡结构如图1所示,其结构参数及相互作用见表2。通过表2可知,从PIP…MPY到PIP…MEK,相互作用能从5.5 kcal·mol-1减少到3.65 kcal·mol-1,复合物趋于不稳定。作用距离由2.929 Å逐渐增加到3.018 Å,其增加趋势与结合能的减小是一致的;且均小于两作用原子的范德化半径之和,因此可以认为复合物的形成是I与N(O)之间相互作用的结果。分子内C-I共价键长度也随之从2.128 Å减小到2.114 Å,其减少趋势也反映了逐渐减弱的分子间相互作用。复合物的作用角度θC-I…B接近180°,表现出明显的方向性,即沿着C-I共价键方向指向N(O)原子,如图1所示。这种直线型作用结构很有可能是由N(O)的孤对电子决定的。

表2 复合物平衡结构参数及相互作用能

2.3 电荷密度拓扑分析
为了解卤键作用的本质,本文分析了平衡结构中分子间I…N(O)接触的电荷密度拓扑性质,发现在三种复合物中,I…N(O)接触都存在着键关键点(图1中I…N(O)接触的(3,-1)关键点),表明I与N(O)原子之间有化学键的存在,这与前面的结论是一致的。关键点处电荷密度拓扑性质列见表3。

表3 I…N(O)作用BCP点处电荷密度拓扑性质 a.u.

2.4 自然键轨道分析
根据NBO理论,复合物的稳定性部分依赖于从N(O)原子的孤对电子轨道到C-I σ*反键轨道之间的电荷转移作用,并由此产生了电荷转移量(Δq)和二阶稳定能(E(2))这两个重要物理量。复合物形成过程N(O)原子的孤对电子轨道与C-I σ*反键轨道之间的轨道作用(nN(O)→σ*C-I)数据列见表4。

表4 三种复合物形成时nN(O)→σ*C-I轨道作用
由表4可以看出,复合物形成时,电荷转移量从26.0 me依次减小到13.0 me,二阶稳定能也从7.9 kcal·mol-1降低到4.0 kcal·mol-1,也说明了分子间相互作用逐渐减弱,这与相互作用能的结果一致。
键长的变化与键上电荷密度密切相关。成键轨道上电荷增加则键长变短,键能变大,键更加稳定;而反键轨道上电荷增加削弱了键的强度,产生相反的结果。复合物形成时,N(O)的孤对电子轨道上的电子大量进入C-I σ*反键轨道,从而使C-I键明显增加,电荷转移量越大,C-I键长增加的越多。
3 结论
1)以高精度MP2计算结果为参考,比较了X3LPY和B3LPY两种泛函,证明B3LYP泛函更适合用来描述卤键体系;2)在结构上,C-I键与给电子体N(O)的作用角度接近180°,表现出明显的方向性;能量上,从PIP…MPY到PIP…MEK,相互作用能逐渐降低;3)复合物中,I…N(O)接触中都存在着(3,-1)关键点,其本质属于闭壳层非共价相互作用;4)复合物形成过程中N(O)原子的孤对电子转移到C-I σ*反键轨道,从PIP…MPY到PIP…MEK过程中,电荷转移量和二阶稳定能逐渐减小,与相互作用能变化一致。
