着色性干皮病一家系
2019-06-01王白鹤周鹏军孙建方
王白鹤 周鹏军 孙建方
临床资料患者,女,37岁。主因“面部发生雀斑样皮损31年”就诊。患者6岁时面部开始出现黑色斑点,并逐渐增多,泛发至全身,以面颈部为著,皮疹日晒后加重。半年前无明显诱因面中部斑点基础上出现一处褐色增生物,约黄豆大,就诊于当地医院,给予手术切除并行组织病理检查诊断“基底细胞癌”,之后未见复发,但面部多发雀斑样皮疹一直未消退,现为进一步治疗来我院,患者自发病以来精神睡眠可,二便正常,体重未见明显变化。体格检查:一般情况较好,各系统检查未见明显异常,发育和智力正常。皮肤科检查:面部及躯干上部、双上肢皮肤干燥,可见泛发性雀斑样皮损,褐色针尖至粟粒大,数目多,部分融合成不规则色素斑,可见白色萎缩斑点、疣状角化及毛细血管扩张。左面部可见小片术后瘢痕(图1a,b)。适龄结婚,育有一儿一女,配偶体健,子女均有类似皮肤表现。患者父母非近亲结婚,其妹妹和兄长亦有相似皮肤病史。家系图谱见(图2)。临床诊断:着色性干皮病。治疗:嘱患者避免日晒,给予遮光剂保护皮肤;同时建议其家属详细体格检查,对出现的早期皮损给予及时保护和预防。
讨论着色性干皮病(xeroderma pigmentosum,XP)是一种常染色体隐性遗传皮肤病。该病在1870年被Kaposi首先报道,它包括7个互补型(XP-A到G)和一个变异型(XP-V)。其中 XPC在美国,欧洲和北非最常见,而XPA是日本最常见的类型[1]。XPV由DNA聚合酶G突变引起,与其他XP类型相比,XPV患者的长期生存率可能增高。
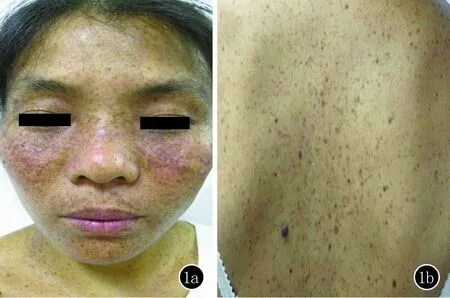
图1a,b 面部及躯干上部、双上肢皮肤干燥,可见泛发雀斑样皮损,褐色针尖至粟粒大,数目多,部分融合成不规则色素斑,可见白色萎缩斑点、疣状角化及毛细血管扩张,左面部可见小片术后瘢痕
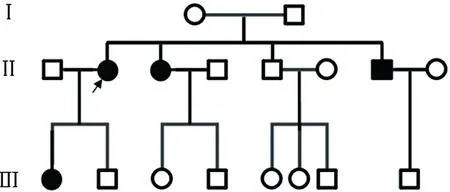
图2 着色性干皮病家系图
XP患者皮肤对日光照射高度敏感。多数患者因为缺乏核酸内切酶,无法修复紫外线照射后皮肤细胞DNA的损伤,常引发抑癌基因突变,因而患者易出现光损伤相关疾病和皮肤恶性肿瘤。此外,20%患者因缺乏DNA聚合酶无法完整修复DNA损伤而致病,如XPV。
XP通常早年发病,起始年龄在6个月~3岁,病变局限于曝光部位,如面颈部、上肢伸侧等,初起轻微日晒伤,随着日光照射时间累积,进行性出现增多的不规则雀斑样皮损,伴色素沉着、毛细血管扩张,皮肤干燥、粗糙,可出现日光性角化病,角化棘皮瘤甚至恶性肿瘤[2,3],如基底细胞癌、鳞状细胞癌,少数情况下继发黑素瘤。据研究报道,XP患者与普通人群相比,出现非黑色素细胞性皮肤癌的风险高达10,000倍,出现黑色素瘤风险是2000倍。这种增加的癌症风险导致XP患者诱发非黑素细胞性皮肤癌年龄缩短58岁,诱发黑色素瘤年龄缩短33岁[1]。PTEN突变是紫外线辐射诱发皮肤变化的重要特征,DNA测序研究显示,PTEN突变率在XP黑色素瘤中高达53%,而在普通人群中突变率只有16%[4]。约75%XP患者可出现眼部损害,如畏光、流泪,结膜炎,睑球粘连,睑外翻以及眼睑肿瘤等。此外20%~30%患者有神经系统疾病和智力障碍,如综合征XP[5]。
该报道患者是一个典型的XP病例。患者皮肤光敏感,色素沉着过度,面部基底细胞癌病史,符合XP的诊断。该病鉴别诊断包括:(1)Peutz-Jeughers综合征(Peutz-Jeughers Syndrome,PJS)又称家族性黏膜皮肤色素沉着胃肠道息肉病,它呈常染色体显性遗传,与STK11基因突变有关,一般在出生时或出生后不久发生,表现为口腔、鼻等腔口部位色素斑点,常有腹痛、腹泻、便血等腹部症状,伴胃肠错构瘤样息肉且易恶变;(2)泛发性黑子:包括发疹性黑子病和多发性黑子综合征。前者发病迅速且较突然,短时间内出现大量黑子,后者常有家族史,属常染色体显性遗传病,表现为多发性黑子伴多种先天性缺陷,如心脏发育异常,肺动脉或主动脉狭窄等;(3)COFS综合征是一种常染色体隐性遗传性疾病,为科卡尼综合征(CS)中病情最为严重的一类。又称先天CS综合征,与三种DNA修复基因CBS、XPD和XPG突变有关。特征为小头畸形,先天性白内障,重度智障,面部异形,关节挛缩。
治疗方面,尽量避免日晒,建议遮光剂保护皮肤,同时做好预防,对患者家族中出现的早期皮损给予及时保护。类维生素A的系统治疗有研究报道可以减少皮肤癌的发生[3]。早期切除癌前期恶性病变对于长期生存很重要,此外使用基因治疗和抗氧化剂来减少氧化损伤的检测疗法可能成为未来治疗的新选择[6]。
