杂化硅胶整体材料研磨法制备混合型高效液相色谱固定相
2019-05-29王照地郭丙倩王世革黄明贤
王照地,张 璐,郭丙倩,王世革,黄明贤
(上海理工大学理学院化学系, 上海 200093)
随着生物学、药学、环境科学等学科的快速发展,对高效液相色谱(HPLC)分离分析方法的要求越来越高。色谱柱是HPLC的核心,不同性能的填料是HPLC广泛应用的基础。以硅胶为基质的固定相是目前应用最为广泛的HPLC填料[1-3]。这主要是因为硅胶微球具有良好的机械强度、合适的孔结构和理想的比表面积、稳定的形貌,以及其微球表面含有丰富的硅羟基可以很方便地进行化学键合和表面修饰[4]。硅胶的颗粒大小、孔径分布及其比表面积等都是影响色谱分离效果的重要因素。常见的硅胶制备方法有硅酸盐酸化法[5]、溶胶-凝胶法[6]、喷雾干燥法[7]、堆砌硅珠法[8]等。硅酸盐酸化法会引入一些金属杂质导致填料具有催化活性和非特异吸附性。溶胶-凝胶法和喷雾干燥法制备硅胶时要求的工艺参数比较复杂,需要对各个工艺参数进行优化选择。堆砌硅珠法制备的硅胶微球孔结构是硅胶微粒的间隙,其孔径受限于硅溶胶胶粒的大小。
硅胶整体材料是以烷氧基硅烷为主要原料,采用溶胶-凝胶工艺制备而成的无机整体材料。硅胶整体材料具有机械强度高,稳定性好,孔隙率高,并且含有丰富的硅羟基,易于化学修饰与键合等优点,因此可被用来做快速分析的HPLC固定相。溶胶-凝胶法是一种制备材料的湿化学方法,这种制备方法可以在低温等工艺条件较温和的情况下,通过化学方法控制和调整材料的微观结构,制得的材料具有高纯度、高均匀性、可重复性等特点。在微柱色谱分析领域,通过溶胶-凝胶工艺在毛细管中形成硅胶整体材料[9]是一个常用的方法;在此过程中可引入不同的有机官能团,它们不参与反应而是直接引入到硅胶表面制得有机-硅胶杂化整体材料[10]。多种硅烷化试剂可被用来制备杂化整体材料,比如辛基三乙氧基硅烷(C8-TES)[11-13]、苯基三乙氧基硅烷(PTES)[14,15]等等。但是,常见的毛细管整体柱[16-18]应用到HPLC分析系统时对色谱仪器有较高的要求。Joung等[19]和Faiz等[20,21]将硅胶整体材料研磨得到弯曲型和分叉型等不规则形状的硅胶颗粒,这些形状的硅胶颗粒相对于传统的球形硅胶颗粒在相近粒径情况下有着柱压低和通透性好等优点,可以实现快速、高效的色谱分离效果。由此,整体柱领域的丰富研究成果就可以应用到一般HPLC填充色谱柱的制备中来。在本工作中,我们完成了一锅法引入有机官能团制备出杂化硅胶整体材料,经球磨机研磨得到粒径大小和比表面积均合适的硅胶颗粒,然后对颗粒表面进行化学修饰制备混合型HPLC固定相等一系列实验研究。
近年来,有关混合模式色谱(mixed-mode chromatography)[22,23]的制备和应用越来越受到重视,其中最常见的是具有正交性质的反相和离子交换的结合[24,25]。Dabre等[26]制备出一种新型的反相/弱阴离子交换固定相,使得多种维生素达到很好的分离效果。彭西甜等[27]利用一种辛基-羧基共同修饰的反相/弱阳离子交换色谱柱,实现了多种表面活性剂、药物、碱性溶质分子混合物的基线分离。王晓欢等[28]制备了一种苯基-苯磺酸共同键合的反相/强阳离子交换混合色谱固定相,应用于几种碱性药物的分离。唐涛等[29]以十八烷基三氯硅烷和3-巯丙基三甲氧基硅烷为原料,制备了一种C18-磺酸基团共同修饰键合的反相/强阳离子交换混合色谱柱填料,实现了多种苯的同系物和若干种核苷的基线分离,还对牛血清蛋白(BSA)的酶解产物实现了有效的分离。混合模式反相弱阳离子交换(RP/WCX)色谱具有疏水和弱阳离子交换的保留机理,结合了两者的优势,可以解决传统反相液相色谱(RPLC)中碱性化合物分离效果差和色谱峰过载的问题。鉴于此,本文制备了一种含C18和羧酸基团的反相和弱阳离子交换(RP/WCX)混合模式色谱固定相,并考察了其反相保留作用能力和对碱性药物的分离作用和保留行为。
1 实验部分
1.1 材料与设备
聚乙二醇(相对分子质量20 000)、四甲氧基硅烷、乙烯基三甲氧基硅烷、三羟甲基氨基甲烷(Tris)、11-巯基十一烷酸、普鲁卡因、利多卡因、普萘洛尔购自上海阿拉丁生化科技股份有限公司;尿素、乙酸、咪唑、甲醇、乙醇、异丙醇等(均为分析纯)购自国药集团化学试剂有限公司;甲苯、三氯甲烷购自上海凌峰化学试剂有限公司;C18(十八烷基二甲基氯硅烷)购自美国Gelest公司;反向测试样品购自大连依利特公司;偶氮二异丁腈购自天津市光复精细化工研究所。
230Ⅱ系列高压液相色谱仪和EC2006色谱数据处理工作站(大连依利特分析仪器有限公司); VEGA3扫描电子显微镜(SEM)和元素能谱分析(EDS)(泰思肯贸易上海有限公司); Tristar Ⅱ 3020比表面与孔径分析仪(美国Micromeritics公司);行星式球磨机(长沙德科仪器设备有限公司);液相色谱装柱机(深圳正大流体机电设备有限公司), KSL箱式高温烧结炉(合肥科晶材料技术有限公司),电热鼓风干燥箱和真空干燥箱购自上海-恒科学仪器有限公司,KQ-300DE型数控超声波清洗器(昆山市超声仪器有限公司),高速台式离心机(德国NUAIRE公司), XL30傅里叶变换红外光谱仪(荷兰Philips公司)。
1.2 实验方法
1.2.1杂化硅胶整体材料的制备
在盛放30 mL 0.01 mol/L乙酸溶液的反应釜内衬中,加入3.24 g聚乙二醇和3.30 g尿素,在0 ℃的冰水浴中搅拌10 min,加入10.0 mL的四甲氧基硅烷和3.3 mL的乙烯基三甲氧基硅烷,继续搅拌40 min;然后将反应釜置于40 ℃的环境下反应48 h,接着在120 ℃的环境中反应48 h,即得到硅胶整体材料。将硅胶整体材料用水和乙醇分别清洗2次,在70 ℃下干燥20 h。
1.2.2硅胶整体材料的研磨
调整球磨机的研磨条件,将打碎的硅胶整体材料研磨2 h后,收集硅胶颗粒,用水、乙醇和甲醇分别离心洗涤2次后在60 ℃真空干燥24 h。
1.2.3硅胶颗粒的Tris处理
称取5.4 g Tris溶于150 mL水中,加入6.0 g研磨离心洗涤干燥后的硅胶颗粒,加入乙酸调节溶液的pH为9;然后将其放在155 ℃干燥箱中反应10 h,待反应完毕后,用水和甲醇分别离心洗涤硅胶颗粒2次,最后在70 ℃真空干燥8 h。
1.2.4表面键合
称取经Tris处理的干燥的SiO2颗粒4.0 g,置于250 mL三颈圆底烧瓶中,并加入120 mL甲苯,于油浴锅中磁力搅拌,并升温至110 ℃使甲苯回流20 min去除烧瓶中的水分,然后加入1.6 g咪唑(Im),搅拌15 min后用滴液漏斗在20 min内加入3.6 g十八烷基二甲基氯硅烷和30 mL甲苯的混合溶液,继续搅拌悬浮液8 h。将反应液冷却至室温,待沉降后,过滤除去清液,加入100 mL甲苯洗涤固体颗粒,依次用45 mL甲醇、甲醇-水(1∶1,体积比)混合液和甲醇离心洗涤。最后将所得颗粒在60 ℃真空干燥12 h,得到表面键合C18的硅胶固定相(ML-ODS)。
取上述键合C18的硅胶颗粒2.0 g,置于250 mL三颈烧瓶中,加入150 mL甲苯,氮气保护下磁力搅拌并加热至70 ℃,然后加入1.0 g 11-巯基十一烷酸和0.35 g偶氮二异丁腈(AIBN),磁力搅拌反应8 h。待反应液冷却至室温并沉降后,过滤除去清液,加入100 mL甲苯洗涤固体颗粒,依次用45 mL甲醇、甲醇-水(1∶1,体积比)混合液和甲醇离心洗涤,最后在60 ℃真空干燥12 h,得到C18和羧酸基团共同修饰的双配体混合模式硅胶固定相(MM-OCS)。
1.2.5色谱柱的装填
采用高压匀浆法装柱。称取适量微球样品(干重1.8 g),超声均匀分散于50 mL异丙醇-三氯甲烷(1∶1, v/v)混合溶液中,在35 MPa的压力下,以甲醇为顶替液,将固定相填充到150 mm×4.6 mm的色谱柱中。
1.2.6色谱条件
反相应用测试的色谱分离条件:流动相为甲醇-水(75∶25, v/v);检测波长为254 nm;流速为0.8 mL/min;柱温为25 ℃。
碱性药物的色谱分离条件:流动相为乙腈-50 mmol/L NaH2PO4(50/50, v/v, pH=7.0);检测波长为220 nm;流速为0.8 mL/min;柱温为25 ℃。
2 结果与讨论
2.1 硅胶颗粒的制备过程和反应机理
本实验以聚乙二醇为致孔剂,四甲氧基硅烷和乙烯基三甲氧基硅烷为硅胶前驱体,在乙酸和尿素加热分解提供的碱性环境催化作用下,硅烷水解缩聚得到杂化硅胶整体材料。经球磨机研磨后得到粒径大小合适的硅胶颗粒,经Tris处理后,得到具有合适孔径的硅胶。
本实验探究了不同硅源的影响。由于四甲氧基硅烷的水解速率大于四乙氧基硅烷,且文献[30]报道以四甲氧基硅烷为硅源,易于形成孔径分布窄、比表面积大的结构。因此常采用四甲氧基硅烷制备硅胶整体材料。以四甲氧基硅烷和甲基三甲氧基硅烷混合物为硅源得到的硅胶杂化整体材料硬度太大,使用球磨机研磨时得到的颗粒大小不均匀,并且经扫描电镜观察到颗粒大小分布范围较广,形状极不规则。以四甲氧基硅烷和巯基三甲氧基硅烷混合物为硅源时,反应时有明显的分层现象,不能形成理想的带有官能团的硅胶整体材料。以四甲氧基硅烷和乙烯基三甲氧基硅烷混合物为硅胶前驱体时,得到的杂化硅胶整体材料具有类似于四甲氧基硅烷单独作为硅胶前驱体时较满意的结果。
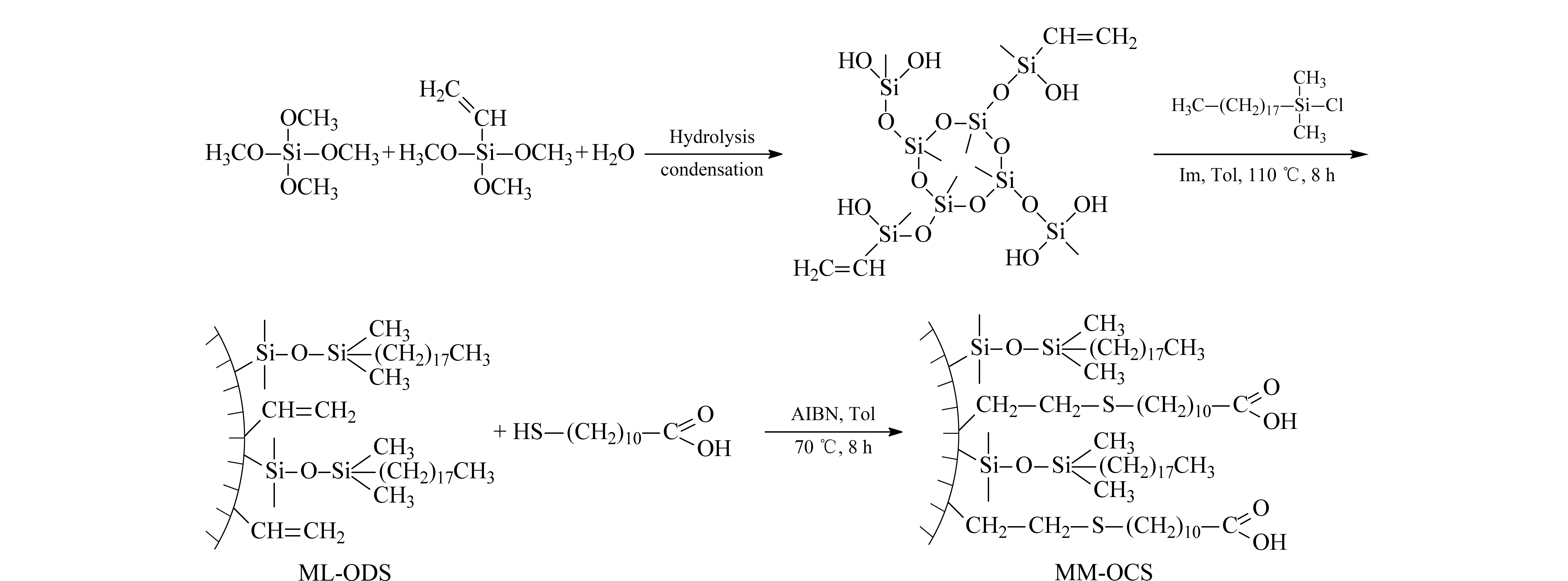
图 1 MM-OCS的制备过程Fig. 1 Schematic procedure for the preparation of MM-OCSIm: imidazole; Tol: toluene; AIBN: azodiisobutyronitrile.
杂化硅胶整体材料的形成过程以及后续的表面修饰可用图1来说明。C18硅烷键合后,再利用硅胶颗粒表面的乙烯基提供的双键与11-巯基十一酸中的巯基发生Thiol-ene点击化学反应[31-33],从而制备了含C18和羧基的固定相MM-OCS。
2.2 固定相的表征
2.2.1扫描电镜
以四甲氧基硅烷和乙烯基三甲氧基硅烷混合物为硅胶前驱体,制备得到的杂化硅胶整体材料经球磨机研磨后可以得到形状比较均匀的无定型颗粒。图2a是整体柱经研磨后,Tris处理前的硅胶;图2b是Tris处理后的硅胶颗粒。Tris处理是为了使硅胶表面更均匀,并使孔径略有增加。因为Tris溶液是弱碱性的,在水热反应条件下,对硅胶有一定的腐蚀和溶解作用,然而Tris溶液又催化硅羟基的缩聚和硅胶的形成,从而引起硅胶表面结构的重整。(reorganization of the silica network)[34]。

图 2 Tris处理(a)前、(b)后硅胶颗粒的SEM照片Fig. 2 Scanning electron microscope (SEM) images of the prepared silica particles (a) before and (b) after Tris treatment
2.2.2元素分析
对固定相MM-OCS制备过程中的各步产物进行了元素分析,结果如表1所示。键合前硅胶含有C元素,表明杂化硅胶含有乙烯基。C18键合后以及硫醇与烯的点击化学(Thiol-ene click chemistry)反应后的固定相中含有的C元素含量增加,并且在固定相MM-OCS中发现S元素,表明十八烷基二甲基氯硅烷和11-巯基十一烷酸均已成功键合到硅胶表面。

表 1 固定相MM-OCS制备过程中各产物的元素分析结果
2.2.3BET测试
不同四甲氧基硅烷和乙烯基三甲氧基硅烷的体积比会造成硅胶孔径和比表面积的不同(见表2)。

表 2 采用不同体积比的四甲氧基硅烷和乙烯基三甲氧基硅烷
由表2可看出,随着乙烯基三甲氧基硅烷用量的增加,所制得的硅胶颗粒粒径增大,由此可以判断乙烯基三甲氧基硅烷使得杂化硅胶整体材料的硅胶骨架的直径变大,并且使其硬度增加。同时,随着乙烯基三甲氧基硅烷量的增加,所得到的硅胶颗粒孔径减小,比表面积增加。当四甲氧基硅烷与乙烯基三甲氧基硅烷比例为3∶1时,孔径大小与比表面积大小与常用的HPLC硅胶基质接近,并且粒径均匀、大小合适,能满足高压液相色谱填料的一般需要。由图3可以看出硅胶颗粒主要孔径变化范围为6~12 nm,但Tris处理后通过对硅胶表面的硅氧键的降解和重排使硅胶孔径有所增加。这些硅胶颗粒的孔径主要来源于原杂化硅胶整体材料的微孔,整体材料的制备方法保证了所得到的硅胶颗粒具有一定的比表面积和相应的键合量,有助于提高色谱柱的分离效果。

图 3 硅胶颗粒的孔径分布曲线Fig. 3 Pore size distribution curves of the silica particles
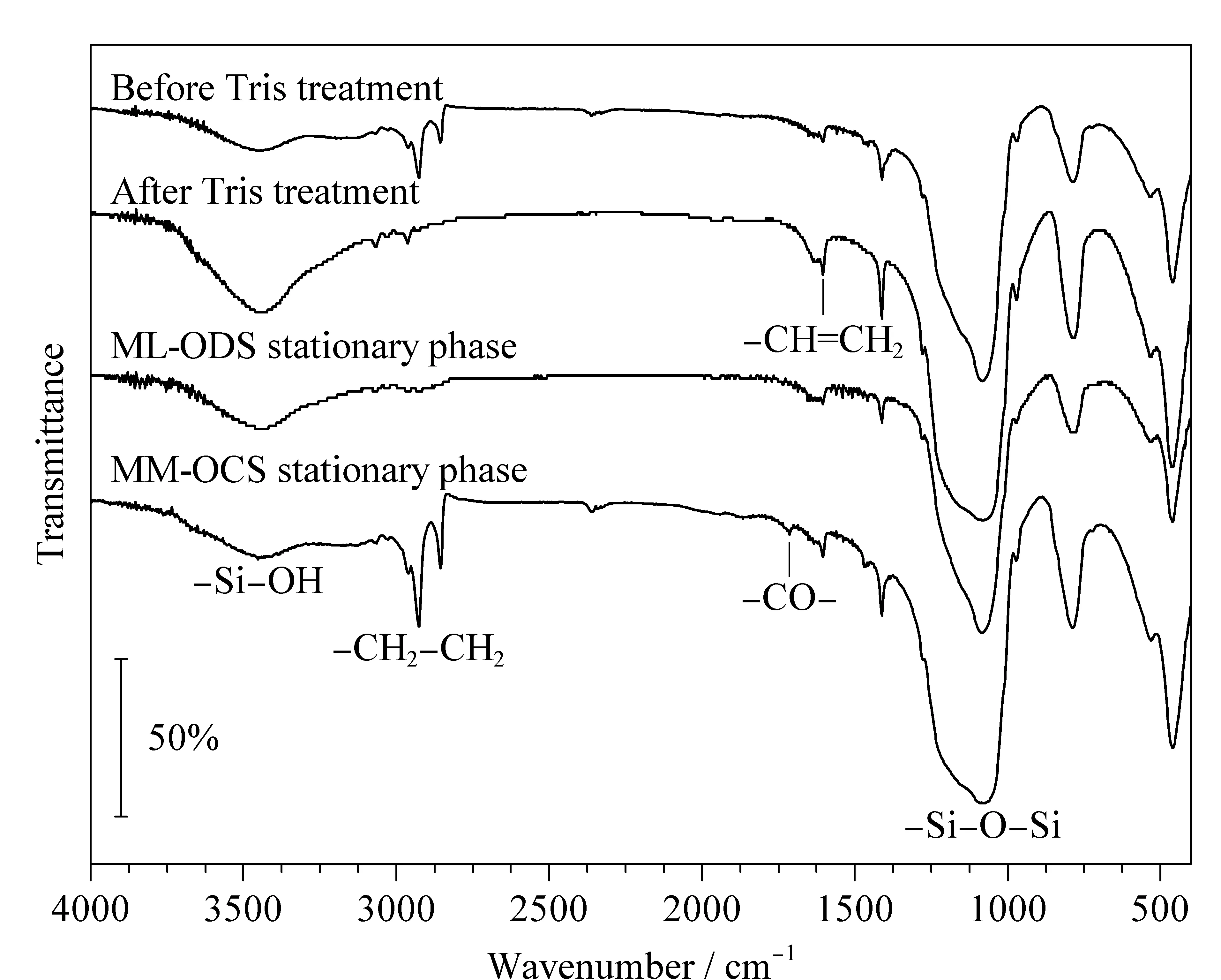
图 4 硅胶颗粒的红外光谱图Fig. 4 FTIR spectra of silica particles
2.2.4红外表征
采用红外光谱对固定相MM-OCS制备过程中的各步产物进行了表征,结果见图4。3 445 cm-1处的吸收峰是残余硅羟基和未充分干燥的水的吸收峰。2 930 cm-1和2 860 cm-1两处的吸收峰分别为亚甲基的不对称与对称收缩吸收峰,这是键合到硅胶表面的十八烷基硅烷与11-巯基十一烷酸中的亚甲基产生的。1 718 cm-1为羰基的伸缩振动峰,1 610 cm-1为碳碳双键的伸缩振动峰,可以推断硅胶颗粒经Tris处理前后,双键仍然保留。1 085 cm-1为硅氧键的伸缩振动峰,并且此处MM-OCS的峰略宽,这是CO-OH与Si-O键振动叠加的结果。以上结果表明,C18和羧基已成功键合到硅胶表面。
2.3 硅胶颗粒的色谱性能评价
2.3.1反相色谱测试
利用混合样品对固定相ML-ODS和MM-OCS进行反相色谱评价。从图5和表3可见,5种化合物得到很好的分离,并且峰形对称。ML-ODS柱和MM-OCS柱是基于同一批键合C18的固定相,但是由图5和表3可以看出ML-ODS柱的反相保留时间比MM-OCS长一些,其原因可能是ML-ODS柱的硅胶表面既有C18又有乙烯基,所以反相保留能力较强;而MM-OCS柱的硅胶表面双键已被修饰成十一烷酸,其所带的羧基使得固定相表面的极性增强,从而减弱其反相保留能力。表3中的理论塔板数(N)和塔板高度(H)与一般相近粒径的硅胶色谱柱相当。
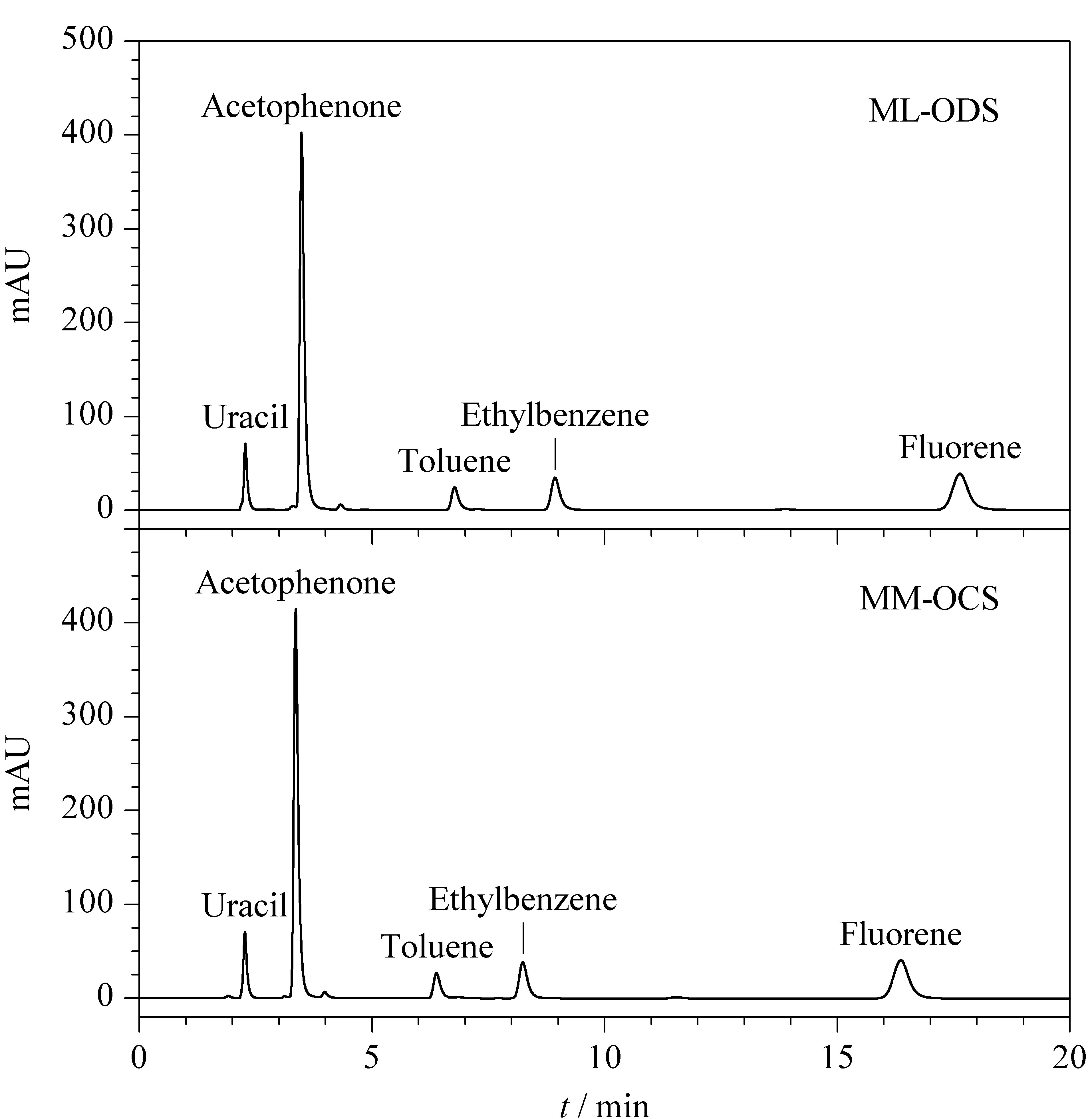
图 5 ML-ODS和MM-OCS在反相模式下分离混合样品的色谱图Fig. 5 Chromatogram of the mixed sample on ML-ODS and MM-OCS stationary phases in reversed-phase mode

表 3 ML-ODS和MM-OCS固定相分离混合样品的色谱参数
tr: retention time;N: theoretical plate number;H: theoretical plate height.
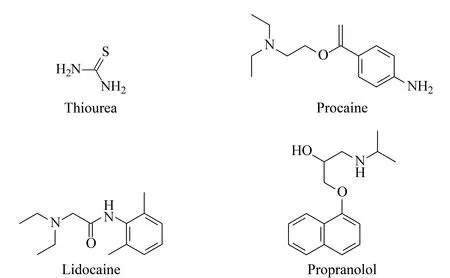
图 7 待测碱性药物的结构Fig. 7 Structures of basic drugs to be tested
由图6的van Deemter曲线可以看出乙苯在ML-ODS和MM-OCS两种柱上的塔板高度随着流速的增加先减小后增加,最后趋于平缓,在0.5 mL/min时达到最小。与一般球型ODS柱相比,这种硅胶材料由于其特殊的形状(弯曲型,分叉型等)使得材料通透性好,在高流速状态下,塔板高度变化不大,因此可以进行快速HPLC分析,体现了此种硅胶颗粒的优势。
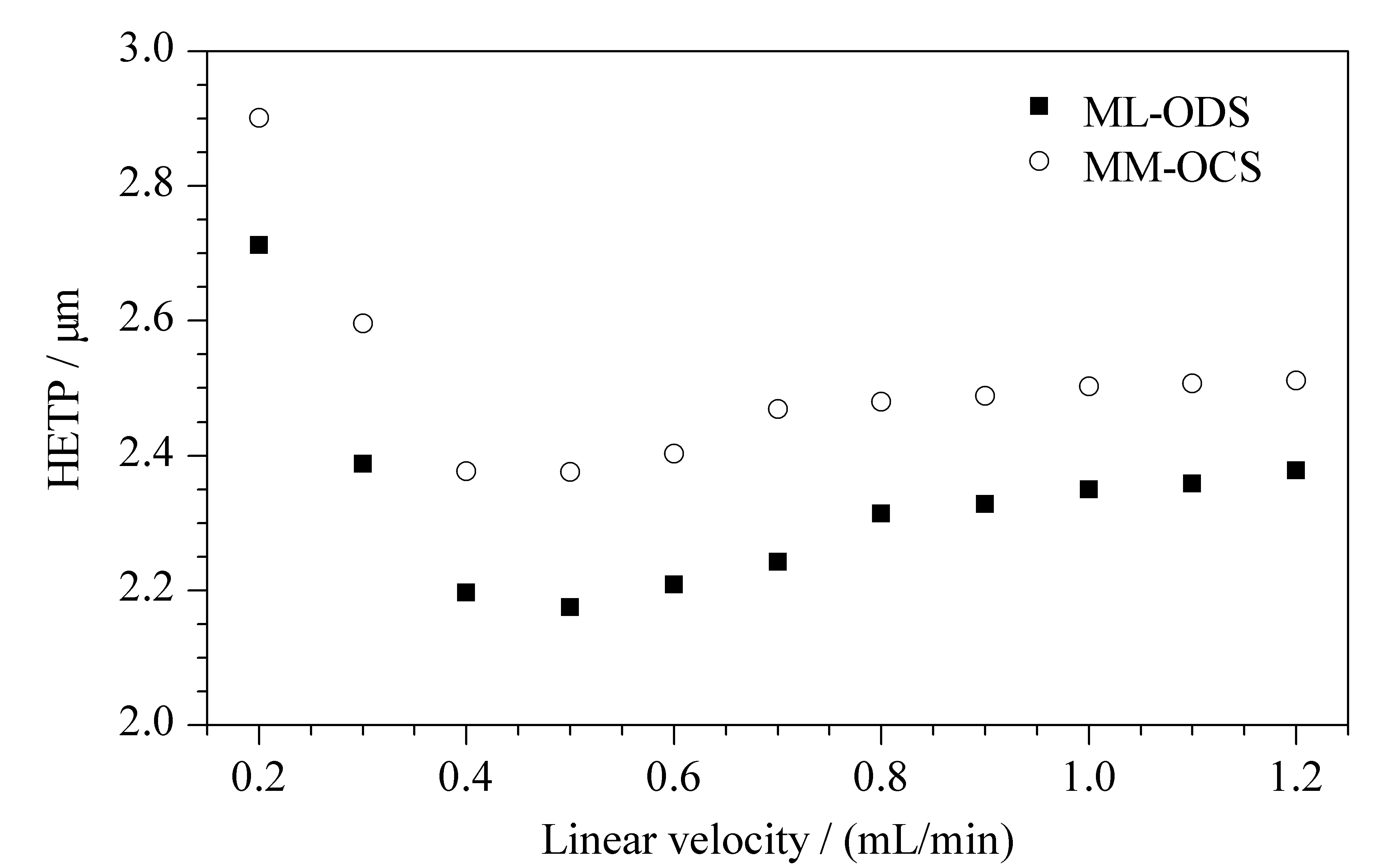
图 6 乙苯在两种色谱柱(150 mm×4.6 mm)上的van Deemter曲线Fig. 6 van Deemter curves of ethylbenzene obtainedon two columns (150 mm×4.6 mm) Mobile phase: MeOH/H2O (75∶25, v/v). Injection volume: 10 μL. Detection wavelength: 254 nm. HETP: height equivalent to a theoretical plate.
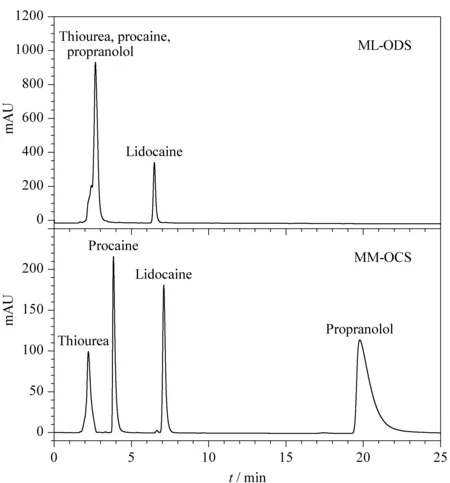
图 8 ML-ODS和MM-OCS分离碱性药物色谱图Fig. 8 Chromatogram of basic drugs on ML-ODS/MM-OCS stationary phase Mobile phases: CAN-50 mmol/L NaH2PO4(50∶50, v/v) at pH 7.0. Peaks: 1. thiourea; 2. procaine; 3. lidocaine; 4. propranolol.
2.3.2离子交换性能测试评价
选取几种碱性药物(见图7)进行分离评价,由图8可以看出在ML-ODS柱上,当流动相处于中性(pH=7)缓冲液条件下时,这几种碱性药物不能被分开,其主要原因是这几种碱性药物的极性或亲水性比较强,它们与固定相ML-ODS的作用弱。但是在MM-OCS柱上,这几种药物却有很好的保留和分离效果,说明混合型固定相中的羧基产生了离子交换作用;同时,碱性比较强的普萘洛尔虽然出现拖尾峰,但是可以在色谱柱上有所保留且可以达到有效的分离。采用前沿色谱的方法使用在ML-ODS柱上不保留而在MM-OCS柱上有保留的碱性普鲁卡因进一步测定了制备的固定相MM-OCS的离子交换当量[35],计算得到普鲁卡因在MM-OCS色谱柱的离子交换当量为560 μg/g。
3 结论
本研究发展了一种利用溶胶-凝胶法得到杂化硅胶整体材料,并通过球磨机研磨和Tris处理,制备均匀、粒径为3 μm左右、孔径在7.5 nm左右硅胶颗粒的方法。以扫描电子显微镜、BET、红外光谱、元素分析等表征手段对所制备的新型硅胶颗粒进行了测试和表征,提供了硅胶制备的优化条件。对所制备的硅胶颗粒进行了C18键合和巯基-烯点击反应得到混合型的硅胶固定相,并对其进行了反相色谱和离子交换色谱性能评价。本研究结果表明这种高压液相色谱填料的制备方法简便,粒径和孔径容易控制,填充柱的通透性好,可以实现快速分析,从而满足不同HPLC分离分析应用的需要和促进HPLC固定相的发展。
