一种高效自动化的转录组差异性表达分析方法
2019-05-16刘振羽王永军邬学敏
赵 芳,高 静,刘振羽,王永军,邬学敏
(内蒙古农业大学计算机与信息工程学院内蒙古自治区农牧业大数据研究与应用重点实验室,中国内蒙古呼和浩特010018)
转录组差异性表达分析是RNA测序数据分析的核心内容,也是转录组研究的基本目标之一。其通过分析不同条件下的转录组数据,进而识别差异基因或异构体,对于揭示基因功能和调控规律具有重要意义[1]。但该过程是一个多步迭代的复杂流程,涉及多软件、多步骤,软件之间还具有复杂的数据依赖关系。传统方式下,研究人员手动执行各环节,前一任务的输出即为后一任务的输入,只有前一任务执行完毕后才可串行执行后续依赖任务[2]。而且,各软件的输入输出格式不尽相同,均需研究人员进行手动的格式转换。该过程中,复杂的命令行操作以及大量的参数配置很大程度上增加了转录组差异性表达分析的难度,导致效率低、出错率高、可重复性差。此外,环境的不易迁移性也进一步导致数据分析问题成为生物信息学的研究瓶颈。
近年来,针对转录组差异性表达分析,人们开发了一些高性能的自动化分析框架,如system-PipeR[3]、SAMSA[4]、miRPursuit[5]。这些框架实现了转录组差异性分析的自动化,一定程度上克服了传统方式需大量手动操作的弊端。但在框架部署与初始配置时,仅提供命令行或特定的编程语言接口,仍需用户进行较多的手动操作,不利于缺乏编程基础的生物信息人员使用。此外,移植性较差,需用户在不同的环境下进行多次重复性部署,且用户无法按需集成新的工具,无法满足研究人员的实际分析需求。
因此,文中针对以上低效重复的手动操作、环境的不可迁移以及无法按需集成等问题,提出了一种基于Docker容器技术的转录组差异性表达分析方法,形成一套可重复、易移植、高效自动化的轻量级分析框架。
1 概念定义与原理
1.1 Docker容器技术
Docker是基于Go语言实现的云开源项目,其主要目标是通过对应用组件的封装、分发、部署、运行等生命周期的管理,使用户的App(Web应用或数据库应用等)及其运行环境能够做到“一次封装,到处运行”。Docker具有部署速度快、开发测试敏捷、提高系统利用率、降低资源成本、跨环境可移植性好等优点。
因此,本研究首次将云开源项目Docker虚拟化技术应用于生物信息学领域,用于解决复杂分析的不可重复性、不易迁移性以及无法按需集成等核心难题,使经验较少或非生物信息专业的研究人员执行复杂的流程分析时就像运行一个简单的生物信息学工具一样。
1.2 最佳实践流程
转录组差异性表达分析流程较为复杂,涉及多步骤的处理,如序列比对、转录本组装、转录本合并等。各处理过程需借助不同的工具软件来实现,但目前各步骤的处理软件层出不穷,软件之间复杂的数据依赖关系使得研究人员往往存在着软件选择难的问题,面临着各软件相互衔接难的困境[6]。
2016年,Ghosh和Cean针对转录组差异性表达分析提出了一套最佳实践流程[7],该流程在提出的几年里被生物信息学领域专家所认可并延续至今仍被广泛使用。因此,文中将该最佳实践流程Bowtie2[8]-TopHat2[9]-Cufflinks[10]-cummeRbund[11]与数据预处理环节相整合,在已构建好参考基因库的前提下,将转录组差异性表达分析主要分为8个处理步骤,形成图1所示完整流程。
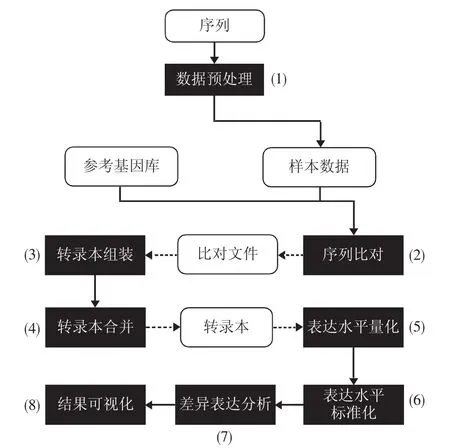
图1 转录组差异性表达分析处理流程Fig.1 The transcriptome differential expression analysis process
1)数据预处理:首先,用SRA-Toolkit(https://hpc.nih.gov/apps/sratoolkit.html)将sra格式的原始测序数据转换为fastq格式;其次,通过FastQC(http://www.bioinformatics.babraham.ac.uk/projects/fastqc)对测序数据进行质量检测;最后,通过Trimmomatic质量检测[12]将质量低或带有接头(a-dapters)的测序数据进行修剪和过滤,进而得到高质量测序数据。
2)序列比对:TopHat2(依赖Bowtie2,比对过程中会调用Bowtie2)将测序数据与参考基因库进行比对,输出结果文件为accepted_hits.bam。
3)转录本组装:Cufflinks将TopHat2/Bowtie2比对结果accepted_hits.bam及参考基因库作为输入文件构建转录本,并计算出各转录本FPKM值。
4)转录本合并:Cuffmerge将多个转录本合并成一套转录本集合,将Cufflinks生成的gtf文件融合为一个更加全面的注释文件merged.gtf。
5)表达水平量化:Cuffquant对单个bam文件表达水平进行定量,用于后续Cuffdiff分析。
6)表达水平标准化:将Cuffquant的输出结果作为Cuffnorm的输入文件,对转录组标准化后的表达水平进行计算,可得到一系列可比较的基因、转录组、CDS组和TSS组的表达值。
7)差异表达分析:通过Cuffdiff计算不同条件下转录本表达水平的显著性差异,进而寻找差异基因。
8)结果可视化:采用cummeRbund软件对Cuffdiff输出结果以热图形式进行可视化呈现,以更直观的方式描述目标差异基因。
转录组差异性表达分析流程中所需软件如表1所示。
2 基于Docker的转录组差异性表达分析方法
2.1 层次构建
基于Docker容器技术的转录组差异性表达分析方法形成的整体框架层次主要分为基础层、核心层、调度层以及应用层(图2)。基础层除硬件设施与操作系统外,还需为上层提供运行环境的Docker引擎;核心层为转录组差异性表达分析容器,属于核心功能组件,可按需集成不同的复杂分析流程;调度层即通过多脚本嵌套触发对服务、资源的协调和控制;应用层即为用户接口层。该框架各层次之间协同配合,进而实现高效自动化的转录组差异性表达分析。
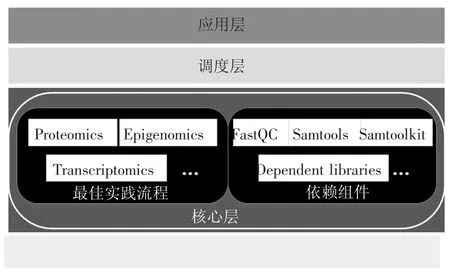
图2 整体框架层次图Fig.2 Framework hierarchy diagram
2.2 核心层
鉴于Docker容器可重复、配置部署快速简单、易于迁移、易于扩展的轻量级特性,文中将1.2节转录组最佳实践流程涉及的软件及其依赖环境封装预配置到Docker容器中,形成一个轻量级自动化的转录组差异性表达分析容器,并将其命名为DEA-container(differential expression analysis container)。容器内部主要通过多脚本联动的方式触发启动DEA-container,控制和协调各组件的执行(核心脚本信息如表2所示),从而实现转录组差异性表达的自动化分析。

表1 最佳实践流程软件列表Table 1 List of best practice softwares

表2 核心脚本列表Table 2 The list of core script
此外,DEA-container还为用户提供了友好的Web图形化界面。我们将全过程封装整合,仅对外提供单一的数据输入和输出端点。用户仅需将目标分析序列提交。全过程无需人工干预,经后台服务器分析完毕后会自动将分析结果文件通过Web界面反馈至用户,供用户下载。
2.3 多脚本联动过程
根据整体的框架构建,各层次之间的协调配合需要通过多脚本联动方式对转录组差异性表达分析的自动化进行实现,具体实现分为以下处理流程,其执行过程如图3所示。
1)数据获取:将用户基于Web图形界面提交的任务及分析参数形成任务表单,采用get_input.sh脚本读取用户数据及相关分析参数并将数据存储于指定位置。
2)初始化检测:通过init_env.sh脚本对宿主主机Docker服务进行状态监测,触发启动DEA-container并对环境进行初始化,主要完成DEA-container配置参数、最佳实践流程组件以及任务数据状态的检测。
3)设置分析参数:对用户提交的任务表单进行分析参数的读取,即分析数据类型,包括分析物种以及物种的名称。通过set_parameters.sh脚本对目标参考基因组、任务数据的存放位置、输入输出参数进行指定。
4)差异性表达分析:参数设置完毕后,通过pipeline.sh脚本触发最佳实践分析流程,分别进行数据预处理、序列比对、转录本组装、转录本合并、差异表达分析等核心处理步骤,最终实现转录组差异性表达的自动化分析,以更直观、专业的热图方式描述差异基因。
5)结果反馈:将结果文件及其热图通过Web图形化界面呈现给用户。同时,为保证研究人员可进行后续研究与分析,通过pull_output.sh脚本对核心结果文件进行提取并以下载列表的形式反馈至用户,方便下载和进行二次处理分析。
以上各环节均存在检错、纠错机制,当某一功能环节检测报错后会选取最近点任务进行断点恢复,这种实时的状态检测与纠错可有效避免任务的中断和失败。在处理过程中,各脚本均会对上一步骤执行状态进行检测,并根据不同的返回值进行相应的操作。若返回值为1,表明上一步骤执行异常,存在错误,程序自动进入纠错环节,将上一步骤产生的结果文件删除后重新执行该步骤,运行完毕会再次检测,检测通过后才可执行下一处理步骤。若返回值为0,则表示上一步骤执行成功,可以顺序执行下一处理步骤。

图3 多脚本联动过程Fig.3 The process of scripts linkage
本研究向用户提供两种服务界面:1)用户图形界面:该方式仅需用户将sra格式的原始序列通过Web用户图形界面进行提交,所需的计算资源由该平台的后台服务器提供,全过程无需用户干预,处理完毕后分析结果会通过Web界面反馈至用户并供用户下载;2)命令行界面:本研究发布了DEA-container以及相关的一键化部署脚本,用户将其下载至本地并通过一键化部署脚本可在任意物理主机基于命令行方式进行特定的数据分析。二者相比,前者更适合于一些经验较少的生物研究人员或非生物信息学研究者进行通用常规的分析,后者更适合于具备丰富的数据分析经验且需进行特定参数分析的研究人员。
3 实验结果与性能分析
为多方面验证DEA-container性能,本研究采用蒙古羊亚系小尾寒羊的高通量测序数据,探讨FSHR基因在不同繁殖力的小尾寒羊中的差异性表达。我们分别选取SRR5786000、SRR5786-001、SRR5786002、SRR5786003 双端数据(共计59.4 GB)作为样本数据集进行分析。样本数据集的详细信息如表3所示。
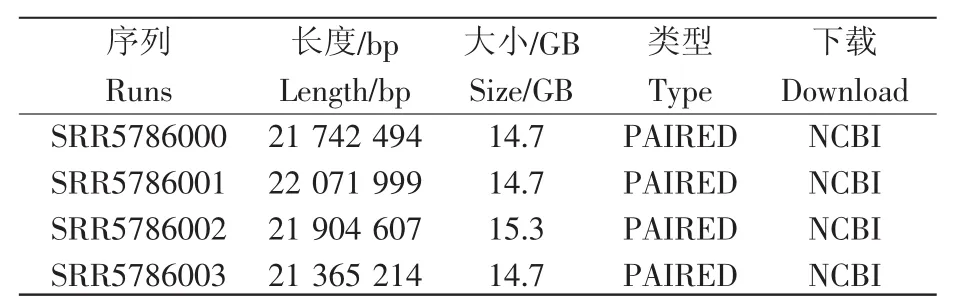
表3 样本数据集Table 3 List of test samples
3.1 准确性验证
准确性是评估一切方法框架最基本的前提。为验证DEA-container框架分析结果的准确性,首先采用两种方式对同一原始数据进行处理分析,即传统人工方式和DEA-container框架全自动方式;随后将两种方式的结果数据进行收集与对比,通过验证结果文件的一致性与完整性来确保数据的准确性。文中主要选取的核心输出文件有TopHat2 结果文件:accepted_hits.bam、Cuffmerge结果文件:merged.gtf、Cuffnorm结果文件:genes.fpkm_table、Cuffdiff结果文件:isoforms.fpkm_tracking。验证方法如图4所示,结果表明不同处理方式所产生的结果文件内容完全一致,证明DEA-container框架具有准确性。

图4 准确性验证方法Fig.4 The method for verifying accuracy
3.2 高效性验证
为测试DEA-container的处理效率,本研究一方面在实验室的惠普工作站(16 GB RAM,4 CPU Intel Xeon)实验平台对小尾寒羊中目标基因的差异性表达分析任务通过Web端进行6次提交和执行;另一方面,采用传统人工方式对相同数据集进行人工的分析处理。我们分别记录了不同处理方式下每个完整分析周期的总运行时间并求取平均值,结果如表4、图5所示。
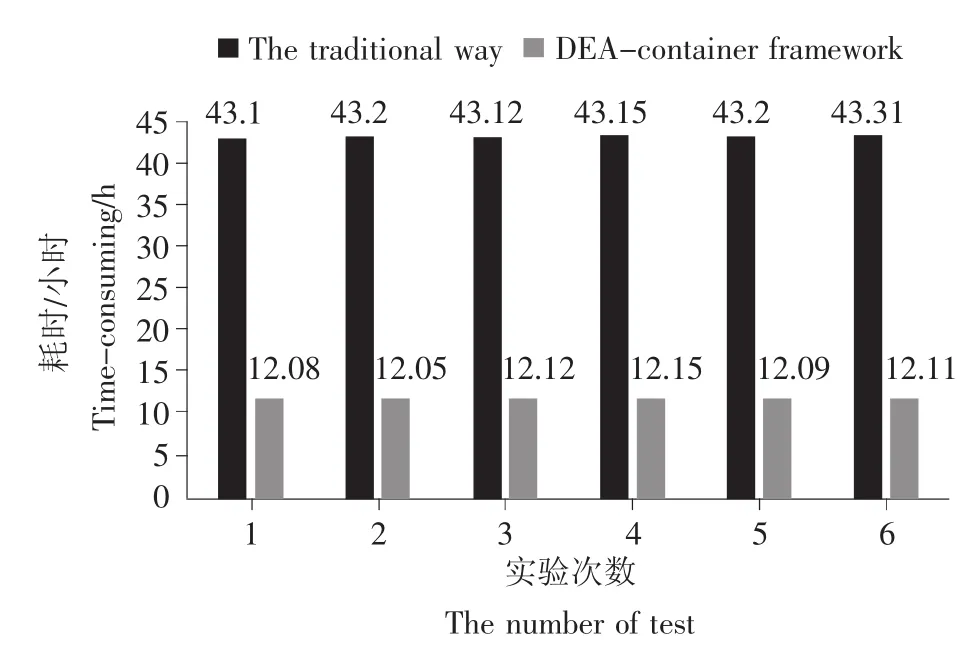
图5 高效性对比验证Fig.5 The comparison of the efficiency

表4 处理时间统计表Table 4 Statistics of the processing time
在传统人工方式处理过程中,TopHat2耗时约 26 h,Cufflinks耗时约 9.9 h,Cuffnorm、Cuffquant、Cuffdiff总耗时约7.4 h,一个完整周期约耗时43 h 18 min。其处理效率差,耗时长,需研究人员手动部署和执行各个处理步骤,工作繁琐且难度大易出错,在大数据时代下该方式不可取[13]。文中采用DEA-container框架全自动化方式进行的6次重复测试均实现了转录组差异性表达分析全流程的自动化执行,对6次执行效率进行计算后取平均值,得到一次完整的分析周期约为12 h 6 min。从以上分析可知,DEA-container的处理效率与传统人工方式相比提升了2倍多,分析时间缩短约72%,很大程度提升了分析效率,在有效降低成本的同时保证了数据分析的高效性。
3.3 部署时间对比验证
DEA-container的突出优势为一次封装、随处运行、可跨平台、不依赖任何编程语言。为了验证这些优势,本研究采用两种方式,即DEA-container一键化脚本部署方式和传统人工方式,将转录组差异性表达分析最佳实践流程部署至11台主机上。但由于传统人工方式基于Linux的命令行,使大量的参数配置以及复杂的命令行操作均受到有无Linux基础的限制,导致部署时间受到较大的影响,因此我们分别选取Linux技能熟练程度不同的人员进行操作,对两种方式的部署时间进行记录,最后求取平均值,结果如图6所示。
由图6可知,传统人工方式的部署时间受Linux技能熟练程度影响较大,其完整部署时间平均约为26.09 h,大量时间耗费在手动的环境配置与软件的部署。与传统人工方式不同,DEA-contianer一键化脚本部署与研究人员Linux技能熟练程度无关,该方式将手动操作的全过程封装且自动化,完整的脚本执行时间平均约26.7 min,高效而自动化,完全解决了软件的环境配置难、安装难等问题,成功地避免了研究人员重复繁琐的手动操作,很大程度上减少了用户工作量、降低了用户工作难度,有效提升了分析效率。同时,DEA-container一键化脚本部署方式降低了对生物研究人员计算机专业技能的要求,更为简单易用。
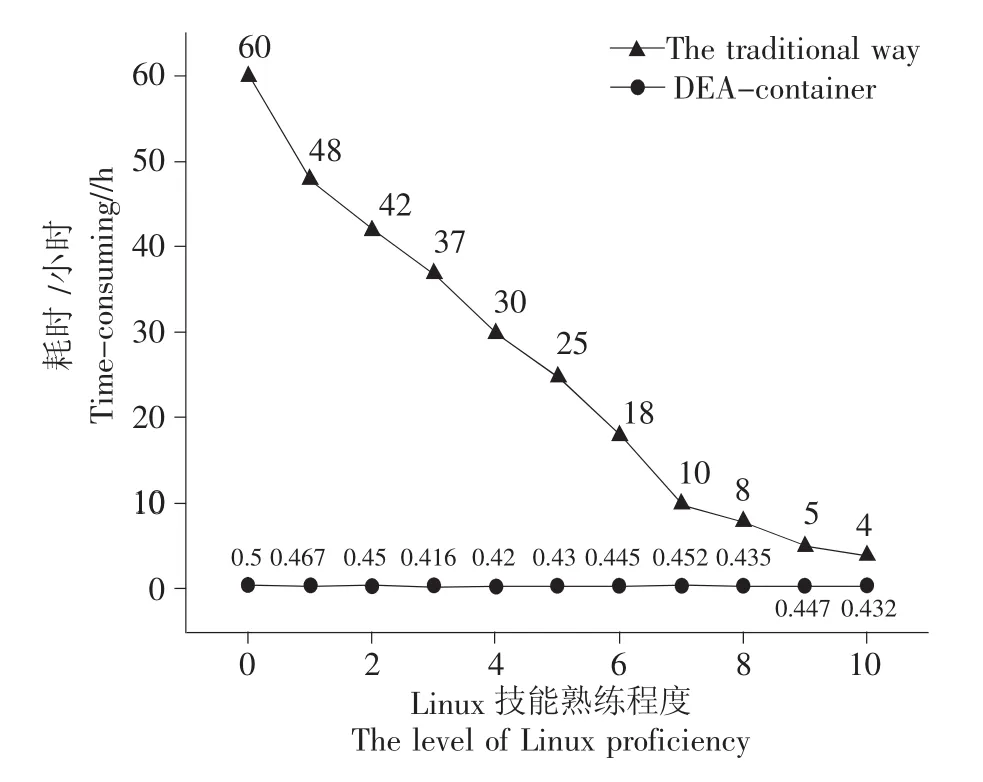
图6 部署时间对比验证Fig.6 The time comparison for deployment
3.4 实验小结
两种处理方式的比对结果如表5所示,与传统人工方式相比,DEA-container方式具有如下优势:1)分析效率显著提升,是传统人工方式的2倍多,分析时间缩短约72%;2)具备较好的移植性,可跨平台随处运行;3)环境部署快速便捷,仅需执行一键化部署脚本即可;4)易扩展,可按需集成多种分析流程与工具软件;5)将复杂流程全面封装,极大简化了用户操作。
4 结语
本文针对转录组差异性表达分析复杂流程形成了一个简单易用的框架,用户只需要通过Web界面对目标分析序列进行提交,无需做其他额外操作,而且后台处理分析完毕后会直接给出分析结果供用户下载,极大地简化了操作流程,提高了分析效率,使得研究人员可以更好地进行数据分析工作,从海量数据中得到更可靠并具有生物学意义的结果。

表5 全局对比Table 5 The global contrast
目前,我们已将转录组差异性表达分析容器及相关内容公布发放至GitHub(https://github.com/fullblossom/the-Differential-expression-analysiscontainer),可供用户下载使用。在后续的工作中我们将按需求形成多种分析容器,并将其公布至平台供用户下载和使用,以期为生物学研究者提供简单易用且高效的分析工具。
