TAF1蛋白及其抑制剂研究进展
2019-05-15陈海芳陈亚东唐伟方
陈海芳,陈亚东,唐伟方
(中国药科大学理学院,江苏 南京 211198)
表观遗传(epigenetics)是指DNA序列未发生变化,但基因表达却发生了可遗传的改变。表观遗传调控机制涉及组蛋白修饰、组蛋白变体、DNA甲基化、非编码RNA修饰及染色质重塑等[1-2],其失调会导致MYC致癌基因过度表达,激活促炎细胞因子,诱导癌症和炎症性疾病进一步恶化[3],是当前生命科学研究中的一个热点。蛋白质的翻译后修饰(post-translational modification,PTM)是表观遗传的重要内容,它建立并维持基因表达的顺序[4-5]。赖氨酸残基的ε-N-乙酰化是蛋白质中最常发生的翻译后修饰之一,常以单个或与其他多种染色质修饰共存的方式,存在于多种核蛋白中,主要是组蛋白[6]。组蛋白的乙酰化修饰通过中和ε-氨基上的正电荷,减弱DNA与组蛋白的相互作用,使本身缠绕紧密的染色质处于松弛状态,从而有助于DNA聚合酶和RNA聚合酶的结合及其他转录因子的招募,促进DNA修复、复制以及基因转录等过程[5,7-8]。组蛋白的乙酰化水平由组蛋白乙酰基转移酶(HATs)、“阅读器”蛋白(reader)和组蛋白去乙酰化酶(HADCs)共同调控[7,9],而这一过程的关键是“reader”蛋白识别能最终决定表型改变的特定PTM。
目前,人们已发现3种组蛋白乙酰化赖氨酸的“reader”蛋白:bromodomain(BRD)[10]、双PHD指蛋白[11]和普列克底物蛋白同源结构域[12]。较长时间内, BRD是唯一的一个为人所知的乙酰化组蛋白“reader”[7]。BRD结构域大约有110个氨基酸组成,在染色质结构变化和转录调节中发挥重要作用。至今,已在人体46种蛋白中发现了61种BRDs,根据其结构和序列相似性分为八大家族[13-14](见图1)。尽管序列变化很大,但所有BRDs共享一个高度保守的二级折叠结构,其由4个反向平行的α-螺旋束(αZ、αA、αB、αC)组成,通过称为ZA环和BC环的柔性环区域连接,它们在序列和电荷上是可变的,并且形成了一个深疏水口袋即乙酰结合位点。尽管各种溴结构域的环区域存在差异,但负责识别乙酰化赖氨酸(Kac)的氨基酸残基是高度保守的。在大多数BRDs中,Kac通过氢键与疏水口袋附近的天冬酰胺(Asn)和酪氨酸(Tyr)残基形成关键的相互作用[15]。

图1 人bromodomain家族树
BRDs作为特异性识别组蛋白乙酰化赖氨酸的结构域,通过募集不同的分子伴侣来调节基因转录[16]。近年来,人们越来越关注开发能够靶向特定BRD的小分子,这不仅有助于阐明含BRD蛋白的生物学功能,还不断开辟了治疗疾病的新领域。TAF1是第VII bromodomain蛋白家族中的重要一员,参与细胞凋亡诱导和细胞周期调控等多种重要的生理过程[17]。TAF1作为转录因子ⅡD(transcription factor ⅡD,TFⅡD)中的重要亚单位[18],通过其串联的bromodomain结构域[分别命名为TAF1(1)和TAF1(2)]识别组蛋白H4双乙酰化的赖氨酸尾部(K8ac/K16ac、K5ac/K12ac、K5ac/K8ac/K12ac/K16ac)[19],结合到核心启动子序列上,启动转录[20](见图2)。TAF1拥有乙酰转移酶活性、泛素激活酶或结合酶活性,在基因转录过程中发挥关键调控作用[17]。TAF1也是组蛋白乙酰转移酶复合物STAGA的组分,可与多种介导复合物协调配合激活转录[21]。而且,作为TFⅡD复合体中的最大组成单位,TAF1还可通过其激酶结构域参与p53的磷酸化,导致p53降解和调控细胞周期G1期进展。此外,TAF1的失活可触发DNA损伤响应[22]。TAF1也是雄激素受体(AR)的一个共激活子,通过其N-末端激酶和泛素激活/缀合酶不同程度地增强雄激素受体转录活性[23]。
虽然TAF1在细胞生长和细胞周期的调节中发挥关键作用,但其与疾病的关系却知之甚少。已知在肿瘤方面,TAF1易受到MYC的致癌活化,导致癌细胞增殖[24]。胃癌和结直肠癌中也发现了TAF1基因的移码突变[17]。TAF1也可作为肝纤维化的标记物,与肝纤维化、肝癌的发生发展关系密切[25-26]。在神经系统疾病方面,TAF1基因突变与一些综合征的发生有关,如X连锁肌张力障碍-帕金森病[XDP(MIM:314250)][27]、RykDax 综合征[28],其主要表现为智力障碍、脸部畸形和神经系统异常[29]。最近有多项研究进一步阐明了TAF1参与XDP的病理过程[30-31],使得TAF1有望成为治疗XDP的新靶点。
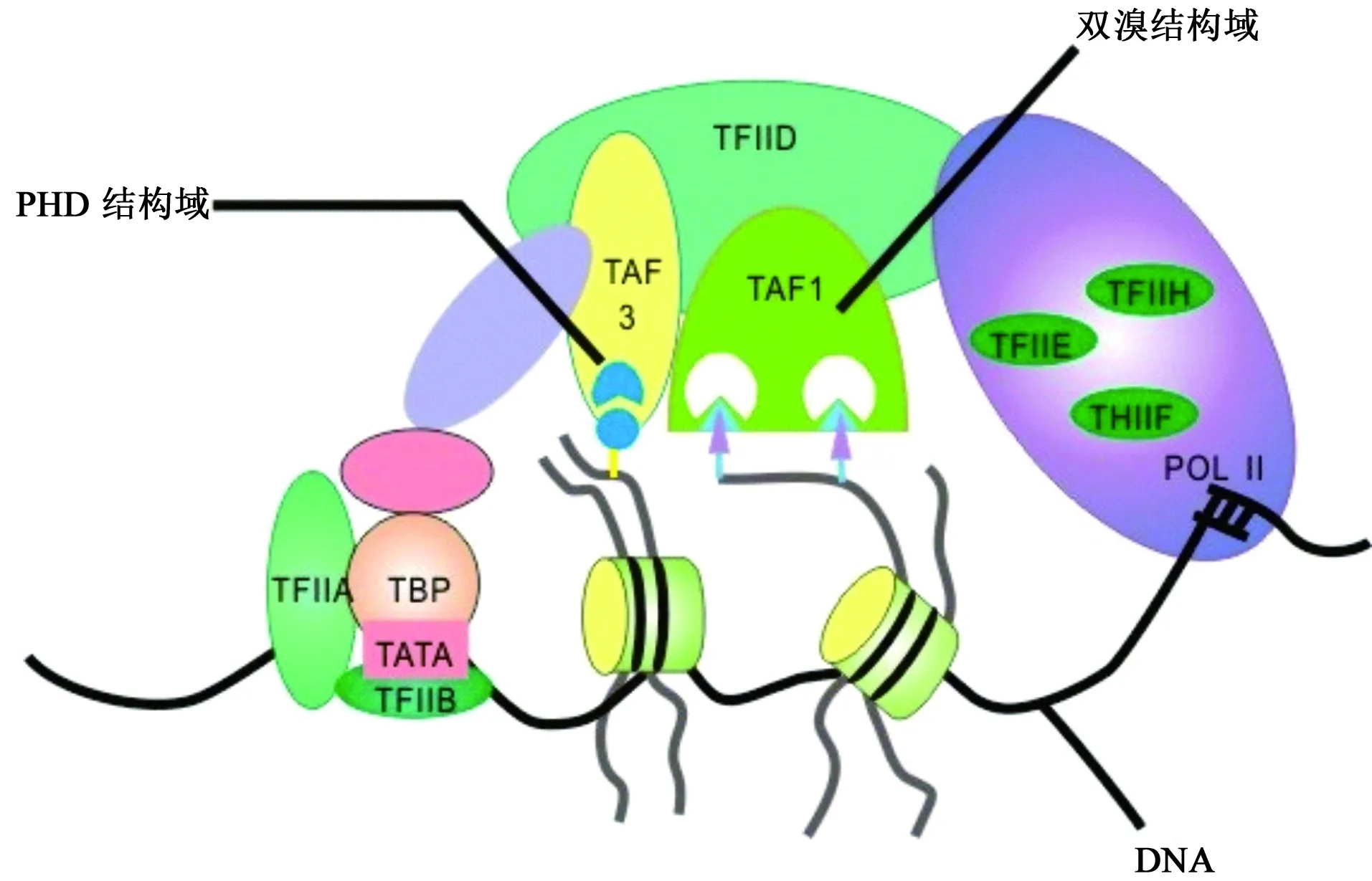
图2 TFⅡD复合体中各结构组成单位与DNA的相互识别作用
TAF1在基因转录过程中是必需的,通过其bromodomain结构域特异性的识别乙酰化的组蛋白,将正转录伸长因子b复合物(positive transcription elongation factor b,p-TEFb)募集至核小体,由此使RNA聚合酶Ⅱ(RNA PolⅡ)的C末端磷酸化且增加邻近基因的转录延伸,介导靶基因的转录激活(见图2)[32]。近期研究进一步表明在转录过程中TAF1可直接与BRD4(BET bromodomain家族成员之一[33])相互作用,协同介导基因的转录控制(见图3A)[21]。在TAF1和BRD4的靶向之间,还存在有强烈的协同作用,使得靶向作用于TAF1 bromodomain结构域的小分子可阻断癌细胞中BRD4介导的MYC转录,从而让TAF1小分子抑制剂对BRD4依赖性癌细胞产生强烈的抑制作用(见图3B),同时,TAF1基因敲除可增加BRD4对癌细胞抑制的敏感性[34]。TAF1与BRD4协同控制癌细胞的增殖,使得TAF1成为一个在由MYC驱动的癌症中具有吸引力的表观遗传学新靶点。
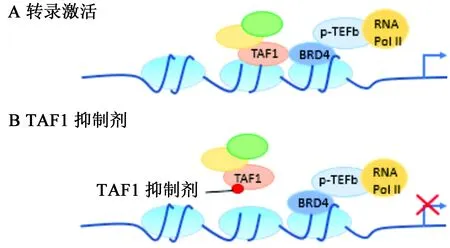
图3 A.TAF1与BRD4基因转录协同调控示意图;B.TAF1抑制对基因转录的影响
在TAF1 bromodomain家族中,TAF1(2)bromodomain因其具有较高的成药性[35],已逐渐成为研究者关注的一个新靶标,目前已报道了多种TAF1(2)bromodomain抑制剂,大致可分为六类:3,5-二甲基异噁唑类、三氮唑类、异喹啉酮类、吡咯并吡啶酮类、苯并异喹啉二酮类和二价TAF1抑制剂。
1 3,5-二甲基异噁唑类
UMB-32(1)[36]是该类母核的一个代表性化合物(见图4)。Bradner课题组使用含氟标记的多组分反应来开发含有3,5-二甲基异噁唑结构的bromodomain抑制剂库。随后对化合物骨架的优化得到了化合物UMB-32,其除了对BRD4有强结合(解离常数Kd:80 nmol·L-1)之外,同时也显示出对TAF1(2)(Kd:560 nmol·L-1)的有效结合。尽管UMB-32与TAF1(2)的结合相对较弱,但这一观察结果表明了一个开发选择性TAF1 bromodomain化学探针的开始。

图4 化合物1的结构及其与TAF1(2)bromodomain的对接(PDB: 5MG2)
2 三氮唑类
Bromosporine(BSP,2)[37]是较早发现的含有三氮唑结构的广谱BRDs抑制剂(见图5),作用于TAF1(2)、BRD2、BRD4、BRD9和CECR2的IC50分别为52、410、290、122和17 nmol·L-1。BSP与TAF1(2)bromodomain的对接显示,其能很好地占据Kac结合口袋。BSP对乳腺癌、白血病、中枢神经系统癌症、前列腺癌、结肠癌等60多种细胞株都有较强抑制,尤其是白血病细胞株。BSP有和泛BET抑制剂(+)-JQ1相似的细胞增殖影响,但抑制活性稍差于(+)-JQ1。
3 异喹啉酮类
Sdelci等[34]在筛选能调节BRD4活性而不直接与BRD4蛋白相互作用的小分子化合物时,发现化合物CeMMEC1(3,见图6)在转录、细胞周期和细胞凋亡中具有与(+)-JQ1相似的细胞效应,且对TAF1(2)显示出一定的亲和力(IC50:900 nmol·L-1)。依托于化合物CeMMEC1与TAF1(2)bromodomain的对接结果,他们继续优化得到了更具选择性的化合物CeMMEC13(4)。CeMMEC13在细胞活力测定中显示出与(+)-JQ1有协同作用。而酰胺连接子反接得到的CeMMEC14(5)却使活性下降很多。这些化合物证明了TAF1(2)bromodomain的成药性,开辟了专门针对MYC诱导的癌症中TAF1(2)bromodomain的新研究思路。

图5 化合物2的结构及其与TAF1(2)bromodomain的对接(PDB: 5MG2)
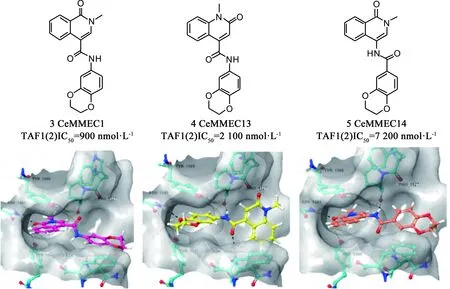
图6 化合物3/4/5的结构及其与TAF1(2)bromodomain的对接(PDB: 5MG2)
4 吡咯并吡啶酮类
2016年,Crawford等[38]发现N-甲基吡咯并吡啶酮结构是一个对BRD家族成员具有强结合效应的片段分子,其对所测试的8个BRD的配体效率(LE)都大于0.54。后续对该片段分子改造得到了化合物6,其作用于TAF1(2)、BRD4(1)、BRD4(2)、BRD9和CECR2的IC50分别为59、92、65、230和240 nmol·L-1,选择性较差。而以1-丁烯基取代甲基的化合物9对TAF1(2)、BRD4(1)、BRD4(2)、BRD9和CECR2的IC50分别为46、2 500、5 500、1 400和4 800 nmol·L-1,选择性较好。化合物9与TAF1(2)(PDB 5I29)的共晶显示(见图7B),吡啶酮的羰基氧及吡咯的氮氢与天冬酰胺1583(N1583)形成二齿的关键氢键,此外吡啶酮的羰基氧还可通过一个水分子与酪氨酸1540(Y1540)形成间接氢键,这很好地模拟了乙酰化赖氨酸和TAF1(2)bromodomain的结合,是该化合物产生活性的主要原因。化合物中的丁烯基相比甲基,能更好地深入附近的空腔,置换空腔内的水分子,诱导形成更稳定的水分子网络,提高了其对TAF1(2)的选择性。此外苯甲酰胺的羰基氧与1533位的天冬酰胺(N1533)的主链氮氢形成直接氢键。

图7 化合物6/9/10的结构及其分别与TAF1(2)bromodomain的晶体结合模式注:图中A、B、C分别是化合物6/9/10在TAF1(2)bromodomain的晶体结合模式,PDB分别为5I29/5I1Q/6DF7
2018年,Crawford等[39]继续对化合物9的甲酰胺和苯环部分进行改造,得到了选择性和活性都更高的化合物GNE371(10),其对TAF1(2)的IC50为10 nmol·L-1,而对其他bromodomain基本上没有活性。化合物GNE371与TAF1(2)bromodomain的共晶结构显示(见图7C),该化合物在保持化合物9原有的氢键作用的基础上,增加了一个咪唑上的氮原子与酪氨酸1589(Y1589)的酚羟基的氢原子之间的氢键作用,这可能是提高选择性的关键。吗啉环取代二甲基后更好地占据了附近的空腔,进一步提高了化合物的活性。在一个测试靶标参与的实验中,即使在100 nmol·L-1的浓度下,化合物10仍然使细胞对(+)-JQ1的敏感性增加。这也是迄今为止,唯一的一个高选择性和高活性的TAF1(2)小分子抑制剂,非常适用于探究TAF1(2)的生物学功能。
5 苯并异喹啉二酮类
Bouchel等[40]在研究选择性的BRPF抑制剂的过程中,意外发现化合物BAY299(11)是一个BRPF2/TAF1(2)双靶点抑制剂,其对BRPF2、TAF1(2)的IC50分别为67、8 nmol·L-1,而对bromodomain其他家族尤其是BET家族,活性较差。BAY299对组蛋白H3、H4与TAF1(2)相互作用的IC50分别为970、1 400 nmol·L-1,而且对血液瘤细胞有较弱的抑制作用。大鼠的药代动力学研究显示该化合物具有良好的药代动力学特性,如低的血液清除率(约占肝血流量的17%),高的稳态分布容积(Vss=1.5 L·kg-1),长的半衰期(t1/2=10 h,静脉注射),良好的生物利用度(F=73%)。BAY299与TAF1(2)的共晶结构显示(见图8),苯并咪唑酮结构模拟乙酰化赖氨酸,与N1583形成关键氢键以及通过水分子与Y1540形成间接氢键。

图8 化合物11的结构及其与TAF1(2)bromodomain的晶体结合模式(PDB: 5MG2)
6 二价TAF1抑制剂
二价结合通过靶向同一蛋白中多个BRD结构域来增强化学探针的亲和力和特异性,是研究含BRD蛋白在疾病中的作用的一种有效手段[41]。2018年3月,Frye课题组[42]报道了一系列针对TAF1 bromodomain的二价抑制剂,包括UNC4495(12)、UNC4512(13)、UNC4928(14)等,这类化合物可以同时结合TAF1中两个不同的bromodomain结构域,其对TAF1 bromodomain复合物的Kd值分别为6.35、10.80和9.69 nmol·L-1。二价抑制剂确实表现出比单价抑制剂更好的细胞性质,这进一步印证了二价结合的有效性,提供了一个更好地探究TAF1蛋白的功能及在疾病中的作用的高效探针。

图9 化合物12~14的结构
TAF1蛋白的表观遗传功能与肿瘤、帕金森病等疾病联系密切,目前研究表明它们有望成为肿瘤治疗的新靶标。TAF1(2)bromodomain已显示与BRD4 bromodomain有协同作用,确定TAF1(2)bromodomain是否也与BET bromodomain家族的其他成员相关联显得尤为必要。如果TAF1只与BRD4有协同作用,那么抑制TAF1(2)bromodomain可以允许选择性调节BRD4活性,这可能会给BRD4抑制的治疗带来益处,有助于避免临床试验中泛BET抑制剂带来的副作用。此外,理解TAF1不同于BET bromodomain的转录效应也可以揭示TAF1抑制剂的替代治疗机会。目前,TAF1(2)抑制剂数量相对较少,母核结构相对单一,获得更多具有高活性、高选择性的TAF1(2)抑制剂来进一步阐明其生物学作用以及与疾病相关作用至关重要。
