红螯螯虾转录组高通量测序及分析
2019-05-15李喜莲郭建林黄振远慎佩晶施伟达顾志敏
李喜莲,郭建林,黄振远,慎佩晶,施伟达,顾志敏
(农业部淡水渔业健康养殖重点实验室/浙江省淡水水产遗传育种重点实验室/浙江省淡水水产研究所,浙江湖州 313001)
红螯螯虾(Cherax quadricarinatus)又被称作澳洲淡水龙虾,外形与海中龙虾十分接近,是一种全球最珍奇的淡水经济虾,澳大利亚为其初始产地。其虾体呈现为褐绿色,有一个膜质鲜红带位于发育成熟的雄虾的螯的外部顶端,十分漂亮,所以又被赋予了红螯螯虾的美称[1]。其不但生长速度快、适应性极强,而且在食物的摄取方面并无特殊偏好,可生存于3~35 ℃的水温中,而且肉质佳、可耐干运,具有较佳的经济效益,它是我们国家引入的第二个淡水品种。I 龄(6月龄)的红螯螯虾一般在秋季上市,而克氏原螯虾主要在夏季,正好填补螯虾市场秋季的空白[2]。
尽管红螯螯虾的经济价值极高,然而其分子生物学探究活动所获取的成果并不乐观,基因数据库的资源也非常少。近年来,高通量测序技术的发展步入高速期,为虾类基因表达的研究贡献了重要支持,不单使测序的时间及成本大大减少,而且能够收获大量的有效数据,对于螯虾生长发育及其抗逆性能等的研究极有帮助。截至现阶段,尚未出现有关于新一代高通量测序技术开展螯虾种质资源创新及开发的资料。
转录组是指生物体的细胞或组织在特定的状态下基因组所转录的全部mRNA,其反映了基因在不同生命阶段、生理状态、组织类型以及环境条件下表达的情况[3]。本研究第一次在红螯螯虾转录组研究活动中选择运用Illumina HiSeq 2000 高通量测序技术,把获取的数据加以拼接及组装,针对所得到的Unigene,参考生物信息学方法剖析基因功能注释和分类、代谢方式等,以功能基因组水平为视角,对红螯螯虾生长发育期间关键基因的表达加以探究,同时也为深层次的分子标记开发及基因功能研究提供有效数据。
1 材料和方法
1.1 转录组测序
试验用的红螯螯虾来源于浙江省淡水水产研究所八里店综合试验基地,选取同一生活环境下的同龄红螯螯虾,分别采集3 个个体的肝脏、精巢和卵巢组织,于-80 ℃超低温冰冻保存备用。采用Illumina TruseqTMRNA sample prep Kit 方法构建文库,使用Illumina HiSeq 2000 进行测序。
1.2 原始测序数据质控
测序接头序列、N 率较高序列、长度过短序列、低质量读段都涵盖在Illumina Hiseq 的原始测序数据当中,这将严重影响后续组装的质量。为了使后期生物信息剖析具有较高的精准度,先对此类数据加以筛选,以使获取的测序数据(clean data)品质较高,从而为后期的剖析活动做好准备,步骤及顺序如下:①将reads 当中的接头序列加以清除,同时将因为接头自连等原有造成未顺利插进片段的reads加以清除;②剪掉序列尾端(3 端)品质较差(质量值小于20)的碱基,倘若余留部分的该数值依旧有低于10 的,那么就需要清除掉整条序列剔除;反之,则留存;③去除含N 比率超过10%的reads;④舍弃adapter 及质量修剪后长度小于70 bp 的序列。
1.3 测序序列拼装
在无参考基因组的转录组的探究方面,在将RNA-seq 高品质测序数据获取之后,需将全部测序读段进行从头组装,以得到单一序列(singleton)以及重叠群(contig),只有完成此剖析之后才能够顺利地继续后期活动。Trinity(http://trinityrnaseq.sourceforge.net/,版本号:trinityrnaseq-r2013-02-25)是目前适用于Illumina 短片段序列组装的一款比较权威的软件,使用该软件对所有clean data 进行从头组装。
1.4 Unigene 功能注释
完成对借助拼接而获取的isogene 序列的注释,同时和string、NR 及gene 数据库加以比较对照。
1.4.1 Nr 注释
借助比较对照,对比所获取序列和NCBI 的数据库(Nr 库),完成提交比较对照的序列的功能标注解释,将对比的数据经由列表展现出来。
1.4.2 GO 注释
GO(gene ontology)是一个数据库,由基因本体论联合会所创立,在物种的类型方面没有过多的倾向性,基本上都比较适合使用,对基因及蛋白功能展开约束及阐述。对其加以应用,能够依据基因参与的生物学经过、结构细胞的具体成分等展开具体的类型划分。所以,GO 注释能够有效助力于基因生物学价值的探究。
1.4.3 COG 注释
比较对照数据库,将COG 注释获取,分类统计全部的基因。
1.4.4 KEGG pathway 注释
KEGG 库(kyoto encyclopedia of genes and genomes数据库)。剖析功能基因的代谢路径,能够更加全面地获悉物种的代谢及合成状况,从而为现实生产活动供应参考。基于国际公认的代谢网络数据库KEGG(http://www.genome.jp/kegg/),对所剖析基因组的基因可能参与全部可能的代谢路径加以供应。
2 结果与分析
2.1 转录组序列的产出、组装
由表1可知,通过Illuminate Hiseq 2000 高 通量测序共获得了147 915 744 条高质量短读序片段,总长度为21 891 279 947 bp,Q 20 值为98.30%,GC 含量占比为40.75%。由Trinity 软件组装,共组装67 369 个Unigene,总长度为69 887 464 bp,平均长度为1 218 bp,N50 长度为1 376 bp。对Unigene 的长度分布特征进行分析可知(图1),在总Unigene 中,0~400 bp 区段所含的Unigene 比例为12.6%,共8 489 个;400~600 bp 区段的Unigene 比例最高为32.38%;601~800 bp 区段的Unigene 占16.26%。经拼接后,共有Unigenes 67 369 个,总长度69 887 464 bp;总isoform 数为93 411 个,从长度为12 690 867.4 bp,平均长度1 037.38bp,最大isoform 长度为34 002 bp(见表2~3,图2)。
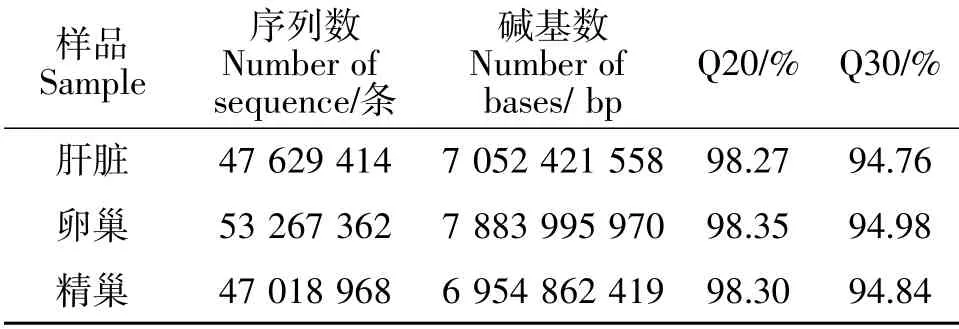
表1 质控后数据量统计Table 1 Data analysis of clean reads
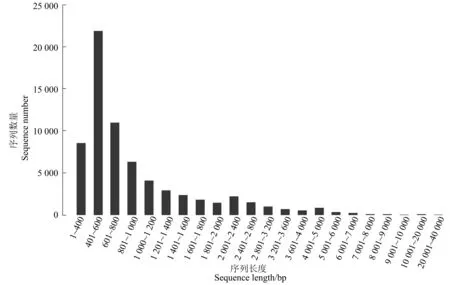
图1 组装序列长度分布Figure 1 Size distribution of transcripts and Unigenes

表2 红螯螯虾转录组拼接结果统计Table 2 Transcript assembly statics for Cherax quadricarinatus

图2 4 个数据库注释的韦恩图Figure 2 Venn diagram annotated on 4 dataset

表3 Mapping 比率统计Table 3 The statistical result of mapping rate
2.2 功能注释
基因注释主要基于蛋白序列比对。比对基因的序列及各个数据库,以将相呼应的功能注释信息所获取。为了剖析工作更加简单,还需整合上述各类信息,以使选出的注释具有最佳的精准度。可以先借助程序来选出比对接近度最高、形式最佳的注释信息,而后在完成少许的人工校对改正。

表4 Unigene 功能注释Table 4 Function annotation of Unigenes
借助BLAST 程序对组装所获取的Unigene 和Nr、GO、COG、KEGG 数据库加以比较,完成Unigene的序列相似性剖析。结果显示,在Nr 数据库当中(见表4),有20 768 个Unigene 能够寻觅到近似序列,约占Unigene 数的30.83%;在GO 数据库当中,有16 989 个Unigene 获取了注释,约为总数的25.22%;在COG 和KEGG 数据库汇中获得注释的Unigene 数量都在1 000 个以下,分别为4 697(占总体数的6.97%)和9 842(占总体数的14.61%)。
2.3 GO 注释
基因本体论(gene ontology,GO)是一个基因功能类型划分数据库,其具有国际标准性,可对各类基因的生物学特点进行较为详尽的阐述。将其运用于红螯螯虾的Unigene 功能类型划分方面,可以将宏观视角上该生物表达基因的功能排列特点加以获取。通过GO 分析(见图3),16 989 个Unigene 被分成了生物学过程(biological process)、细胞组分(cellular component)和分子功能(molecular function)3 个主要类别。
在“生物学过程”当中,就Unigene 数目而言,代谢过程(1 389 个Unigene)功能组居于首位;在“细胞组分”类别中,细胞(762 个Unigene)和细胞部分(761 个Unigene)功能组所含Unigene 数量最多;在“分子功能”类别中,催化活性(1 308 个Unigene)所含Unigene 数量最多。
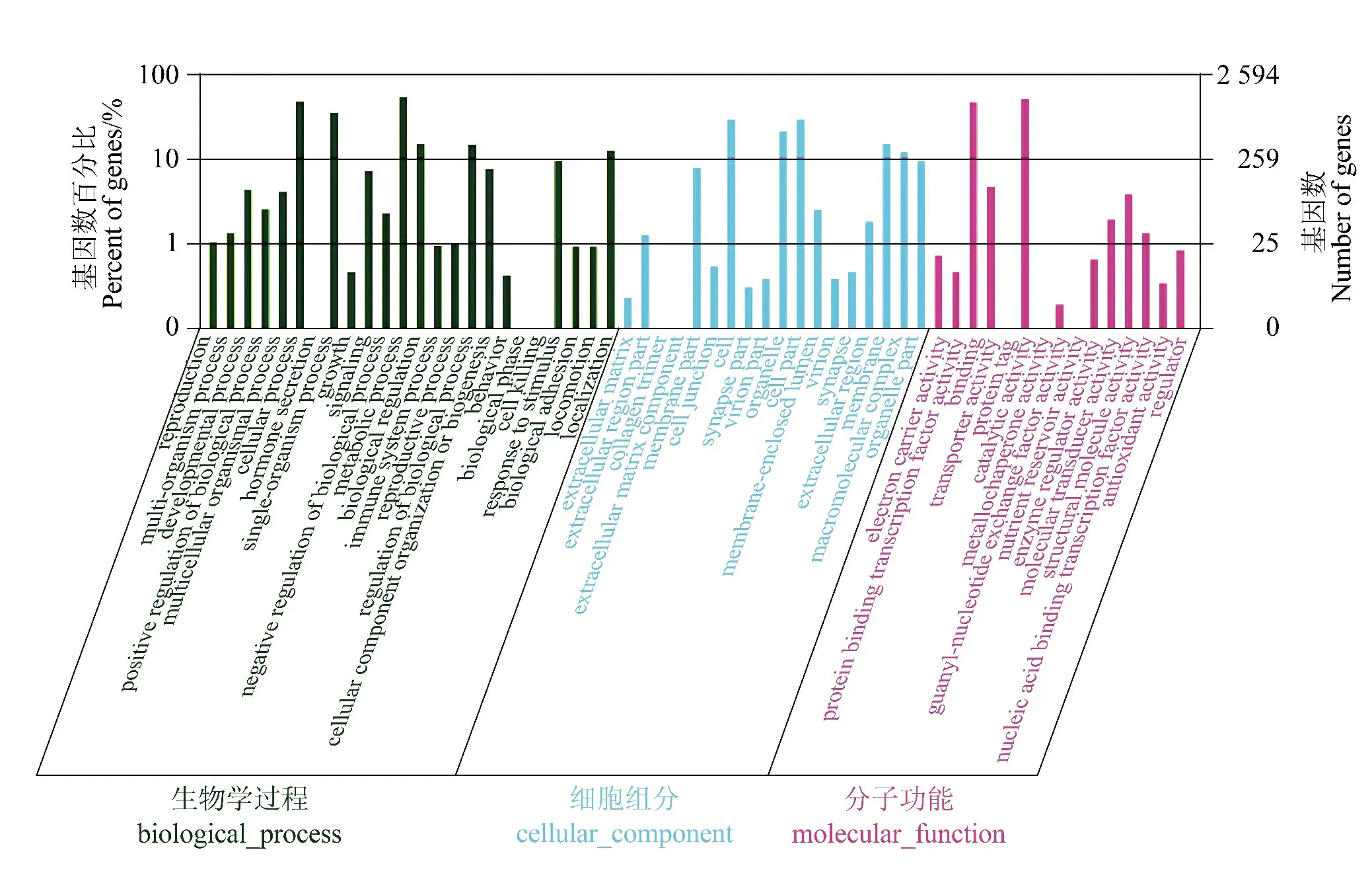
图3 Unigenes 的GO 功能分类Figure 3 Gene ontology (GO)classification of Unigene
2.4 COG 注释
COG 数据库通常运用于同源蛋白注释,由NCBI所研发。其按照蛋白质序列的近似性将后者划分成多个不一样的类,并分别给予特定的COG 编号,用以对一种同源蛋白加以表示。同时,将所有的同源蛋白再分成25 个大类。为了对Unigene 的整体度及注释的有效性展开深度判定,对67 369 个Unigene 加以COG 注释及类型划分,共获得24 个种类,真核细胞的细胞外结构(W)在Unigene 中存在数为0。在24 个COG 注释中,一般功能预测(R)为最大类,共有809个Unigene;然后是转录(K),共有380 个Unigene;原子核功能为最小类,只有1 个Unigene(见图4)。

图4 COG 数据库分类与功能注释Figure 4 Classification and function in COG dataset
2.5 KEGG 注释
KEGG 是一个对基因组、化学以及系统功能信息加以整理合并的数据库。其最为突出的特点即为将已完成整体测序的基因组中所获取的基因目录关联于等级更高的细胞、物种及生态系统水准的系统功能。为了识别红螯螯虾中活性高的代谢通路,对67 369 个Unigene 进行KEGG 代谢途径分析(见图5),将其根据参与的KEGG 代谢通路分为5 个分支:细胞过程(A,cellular processes,4 650 个Unigene),环境信息处理(B,environmental information processing,1 412 个Unigene),遗传信息处理(C,geneticinformation processing,1 207 个Unigene),代谢(D,metabolism,1351 个Unigene),有机系统(E,organismal systems,2 183 个Unigene)。331 个通路当中排列了9 842 个Unigene,而代谢方式所涵盖的Unigene 数目达到了1 534,居于首位(见图6)。
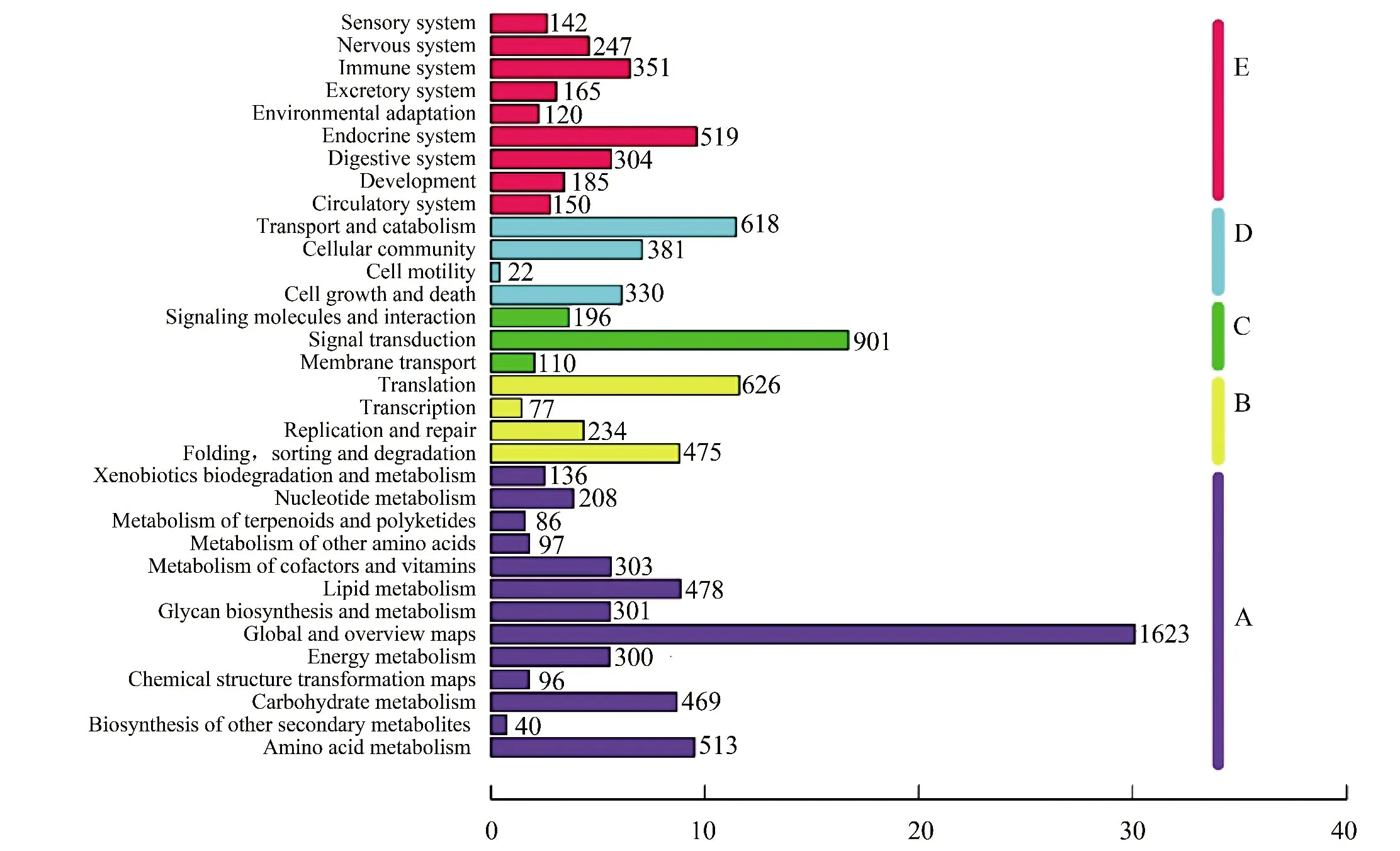
图5 Unigene 功能注释Figure 5 Function of Unigene annotation
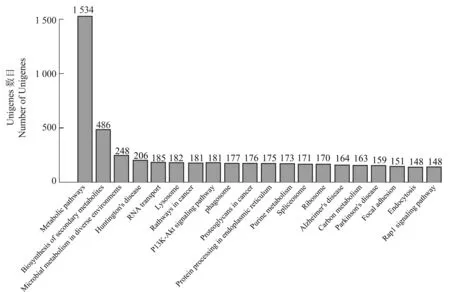
图6 显著富集的KEGG 通路Figure 6 Significantly enriched KEGG terms
3 讨论与结论
近几年,全新一代高通量测序技术被多个领域所运用,而有关于动物基因组的研究也因此而获取了突破性成果。转录组技术在虾类的研究上应用广泛,用于虾类微卫星的筛选[4]、特异相关基因的发掘[5-9]、发育生物学[10-11]等方面。转录组测序的优势在于:①对检测转录本量无上限要求,既可以检测单个碱基差异也可以检测不同转录本的表达。②相比传统微阵列杂交,RNA-Seq 不存在背景噪音问题,信号覆盖动态变化范围大。③高灵敏度,能够检测到样品中只有与几个bp 的稀有转录本,同时能检测到新的转录本,发现未知基因。④无须参考基因组,可分析任意物种的转录组信息[12]。转录组技术的发展大大推进了虾类分子水平的研究。然而与红螯螯虾基因组相关的探究数据却极为少见。Illumina高通量测序不但数据数量庞大、速度较高,而且实效性强、经济性佳,在该物种转录组测序探究活动中尤为适合使用。基于功能基因组学研究当中转录组学的关键性,本研究借助上述技术完成红螯螯虾转录组的测序,以对其基因表达谱加以深层次探究,同时对其生长发展期间的关键表达基因进行发掘。
近年来,高通量测序技术的发展和成熟为各种分子标记的开发提供了大量的资源,加快了各种引物开发的效率,并能达到批量开发分子标记的目标。本研究通过SSR 位点查找发现了单核苷酸重复(>11)位点11 673 个,双碱基重复(>6)位点5 822个,三碱基重复(>5)位点4891 个,四碱基重复(>5)位点290 个,五碱基重复(>5)位点25 个,六碱基重复(>5)位点26 个。本研究还从肝脏、精巢和卵巢组织转录组数据中获得SNP 位点20 654 个(6 097+7 469+7 088),缺失位点12 343 个(3 654+4 437+4 252),插入位点5 611 个(2 443+ 3 032+2 836)。这些分子标记的获得,为开发红螯螯虾遗传多样性分析奠定了分子标记基础,同时也为红螯螯虾QTL 定位、遗传结构分析及基因克隆等研究提供了有效的理论基础。本研究是国内第一次借助Illumina HiSeq 2000 高通量测序技术创建的红螯螯虾转录组数据库,得到了庞大的转录本资料,同时剖析了表达基因的序列组装、功能注释以及代谢路径,为后续的深度研究供应了有效的数据支持,并且上述转录组信息还能够当作后期该物种基因组的参照序列,为此物种的分子生物学研究供应了有较高价值的数据。
