基于溴代比色法的高矿化度浸出液中铀分析方法的优化
2019-05-14张春艳李喜龙阮志龙
张春艳,李喜龙,阮志龙
(中核通辽铀业有限责任公司,内蒙古 通辽 028000)
1铀的测定
1.1 方法原理
在pH 7.7~8.6并含F-条件下,铀与2-(5-溴-2-吡啶偶氮)-5-(二乙氨基)-苯酚(以下简称Br-PADAP)以及F-生成稳定的有色配合物,借此进行比色测定铀[2]。
1.2 试验试剂
除另有说明外,试验过程中所用试剂均为分析纯,试剂和蒸馏水等均符合国家有关标准。
试验用溶液:铀标准溶液,ρ(U)=100 μg/mL(GBW(E)080173);Br-PADAP乙醇溶液,质量浓度为0.5 g/L;酚酞指示剂,质量浓度为1.0 g/L酒精溶液;pH=2酸化水;铀标准储备液,ρ(U)=1 μg/mL;氢氧化钠溶液,浓度为1 mol/L;盐酸溶液,浓度为1 mol/L。
1.3 试验仪器
7230G分光光度计,上海精密科学仪器有限公司;pHS-3E,上海仪电科学仪器股份有限公司;电子天平,精度0.001 g,上海卓精电子仪器股份有限公司。
1.4 分析步骤
1)标准曲线的绘制。准确移取0.00、1.00、2.00、4.00、6.00、8.00、10.00 mL标准储备液,分别置于25 mL容量瓶中,用蒸馏水稀释至10 mL左右。分别加入2 mL混合掩蔽剂,用酚酞作指示剂,用浓度为1 mol/L氢氧化钠和1 mol/L盐酸溶液调酸度至酚酞变无色。然后分别加入2 mL缓冲液、5 mL丙酮、1 mL质量浓度为0.5 g/L Br-PADAP溶液,用蒸馏水稀释至刻度,摇匀。放置20 min后,在波长为565 nm下,用3 cm比色皿,以试剂空白做参比,测量吸光度。以标准液中铀质量为横坐标,吸光度为纵坐标,绘制标准曲线,如图1所示。
由图1看出:标准曲线线性良好,线性相关系数R2>0.999,只是曲线末端1.00μg数据点有点偏离直线。
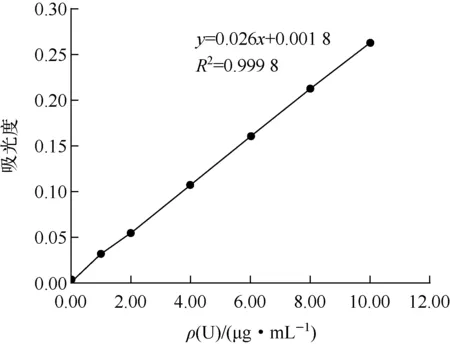
图1 标准曲线
2)样品分析。取适量井场、水冶工艺试样(不超过10 mL),置于25 mL容量瓶中,以下按标准曲线绘制步骤进行操作。
1.5 结果计算
试样中铀含量按下式计算:
ρ(U)=m/V
(1)
式中:ρ(U)-铀质量浓度,μg/mL;m-曲线上查得的铀质量,μg;V-取样体积数,mL。
2溴代比色法的测定下限
溴代比色法用分光光度计制作标准曲线,在一定浓度范围内,溶液中的铀浓度与仪器上响应的信号值呈直线关系,结果可以直接从曲线上查到[3]。在标准曲线上,接近原点的这段是该方法的检出限,但该方法的测定下限在末端的哪个点上、是否从原点开始整条曲线都成立没有做过系统分析。为确定该方法的测定下限,对标准曲线0~1 μg/mL段分段绘制,通过对分析数据的统计分析确定该方法的测定下限[4]。
分别量取ρ(U)=1 μg/mL铀标准储备液0.10、0.20、0.40、0.60、0.80、1.00 mL置于25 mL容量瓶中,按分析步骤绘制标准工作曲线,平行分析4次,试验结果如图2所示。
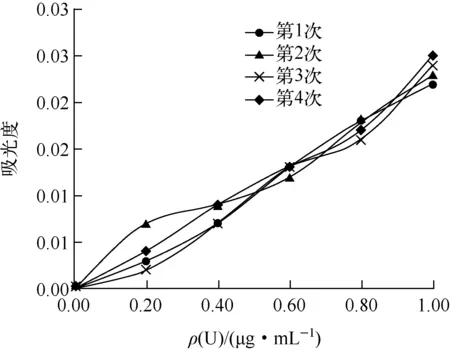
图2 待测液中铀质量为0.10~1.00 μg/mL时标准工作曲线
由图2可见:ρ(U)为0.10~1.00 μg/mL时,曲线中0.00~0.60 μg/mL段吸光度离散程度较大,说明此区间段铀在仪器上响应的信号值不稳定,显色灵敏度不高[5]。为确定该方法的方法测定下限,分别量取ρ(U)=1 μg/mL铀标准储备液0.10、0.20、0.40、0.60、0.80、1.00 mL各12份进行平行测定,结果见表1。
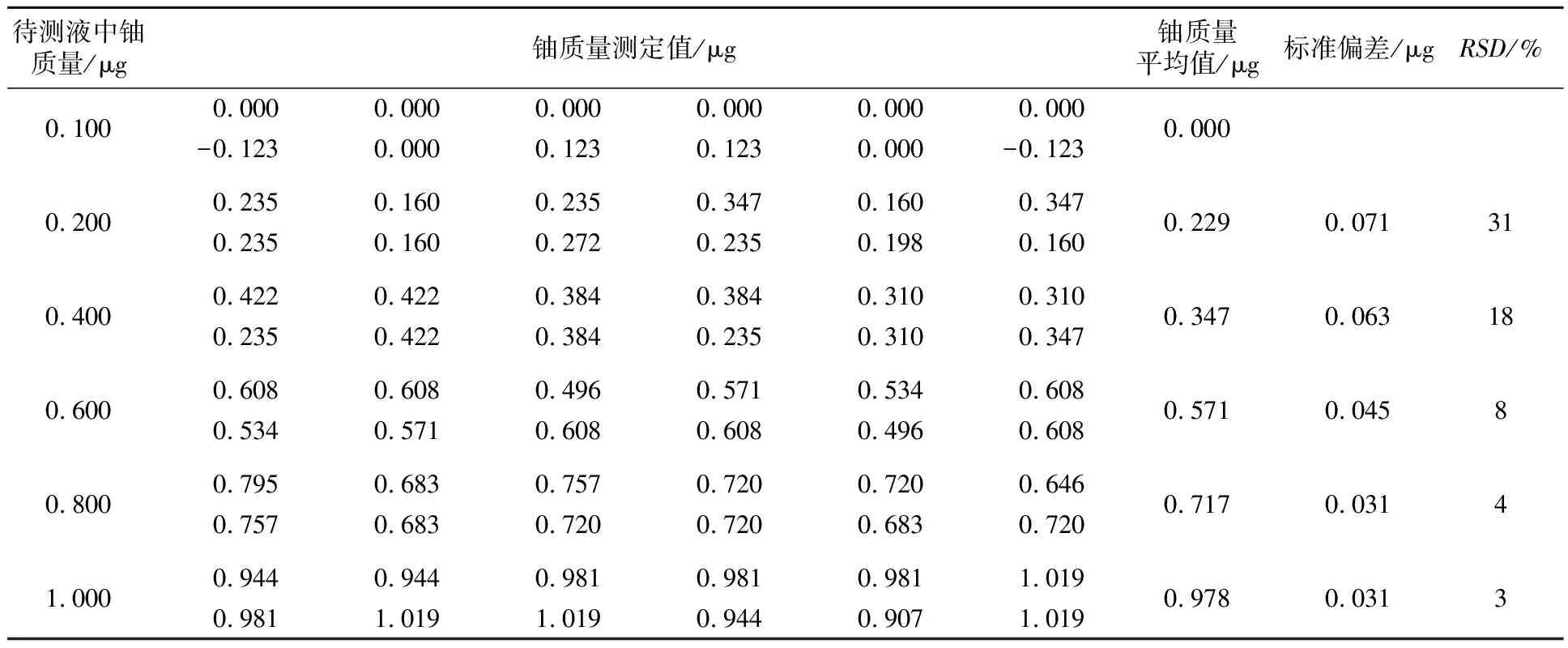
表1 测定方法测定下限
由表1可见,铀质量浓度为0.100 μg/mL时,测定值集中趋势是零,说明铀质量浓度为0.100 μg/mL时不能检出。铀质量浓度为0.200 μg/mL时,测定值标准偏差是0.071,相对标准偏差是31%,数据离散程度较大,测定数据不可靠。铀质量浓度溶液在分光光度计上已有明显的信号反应,因此0.200 μg/mL是该方法的检出限,在检出限附近的测定值只有定性分析的意义,定量测定系统误差值较大。当铀质量浓度分别为0.400、0.600、0.800 μg/mL时标准偏差逐步减小;但0.400、0.600、0.800 μg/mL三个点的平行测定值极差很大,偏离曲线的趋势线较远,不能作为方法的测定下限;而铀质量浓度为1.000 μg/mL的测定值集中趋势是比较明显,平均值为0.978 μg/mL,相对标准偏差为3%,比较接近真值,因此,确定铀质量浓度为1.000 μg/mL是溴代比色法的测定下限。
3干扰因素分析
在确定方法测定下限时,所用的铀标准溶液中不含任何样品基体中的离子,而实际样品的基体离子是很复杂的,为此进行了样品基体所含的离子是否干扰测定以及如何消除干扰因素等试验。
3.1 样品的平行测定
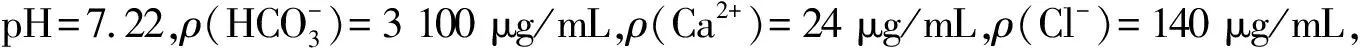
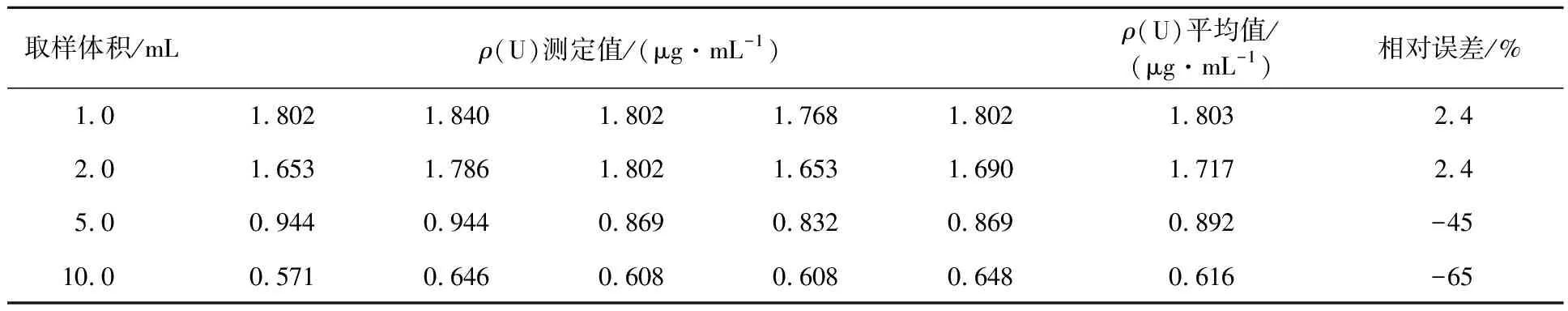
表2 1#浸出液分析结果
注:以取样体积为1.0 mL和2.0 mL时的平均测定值为1#浸出液的铀浓度。
由表2可见:以1#浸出液为例,取样体积为1.0 mL和2.0 mL时,测定值的平均值的相对误差为2.4%,测定值可取;当取样体积是5.0 mL时,相对误差为-45%;当取样体积是10.0 mL时,相对误差为-65%,呈负增长趋势。
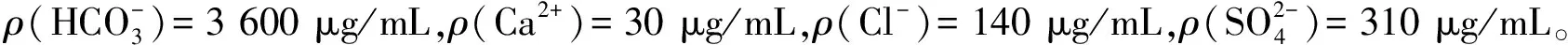
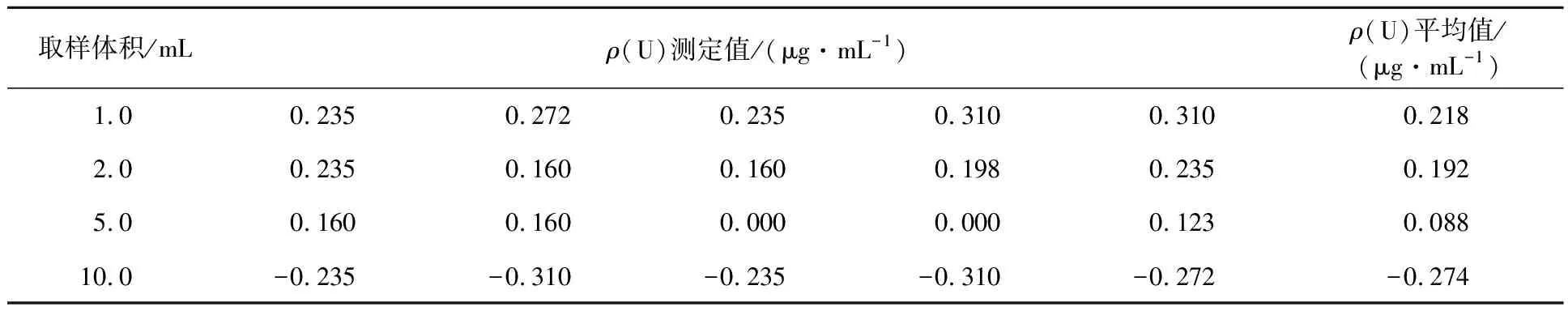
表3 2#浸出尾液分析结果
由表3可见:当样品中铀含量低于方法检出限,取样体积为10.0 mL时,其测定值低于试剂空白值。
试验中除取样体积之外,其他分析条件没有变化,随着取样体积的增大,样品中所含有的杂项离子的量也在增大,而此时铀的测定值不升反降,呈负增长趋势,说明样品固有的基体离子干扰了该方法的测定。
3.2 基体离子干扰测定
基于上述情况,模拟1#浸出液的主要成分,分析测定基体离子干扰因素。当被测元素回收率在90%~110%时,认为基本离子不干扰分析测定。
3.2.1Ca2+的干扰测定
量取7份ρ(U)=1 μg/mL的铀标准储备液各2 mL,分别加入ρ(Ca2+)=30 μg/mL溶液 0.0、1.0、2.0、4.0、6.0、8.0、10.0 mL于25 mL容量瓶中定容,测量加入不同钙质量浓度对铀分析的影响,试验结果见表4。
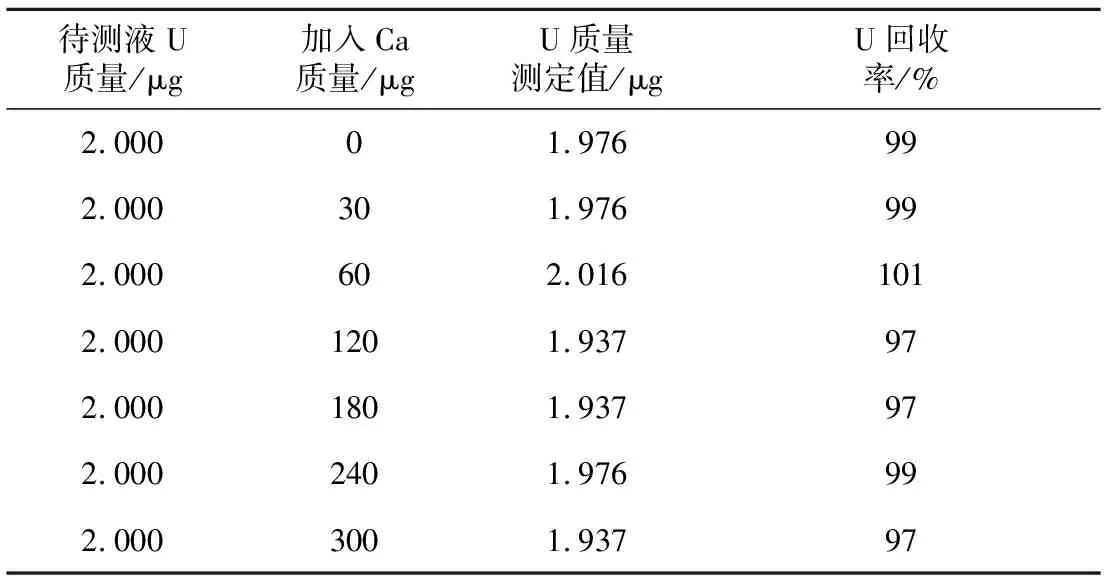
表4 Ca2+对铀的干扰试验结果
从表4可看出,当样品中Ca加入质量在30~300 μg时,即加入钙质量浓度在12 μg/mL的范围内,U回收率在97%~101%,不干扰U的分析测定。
3.2.2Cl-的干扰测定
量取7份ρ(U)=1 μg/mL的铀标准储备液各2 mL,分别加入ρ(Cl-)=200 μg/mL溶液0.0、1.0、2.0、4.0、6.0、8.0、10.0 mL于25 mL容量瓶中定容,考察Cl-对铀的分析影响,结果见表5。
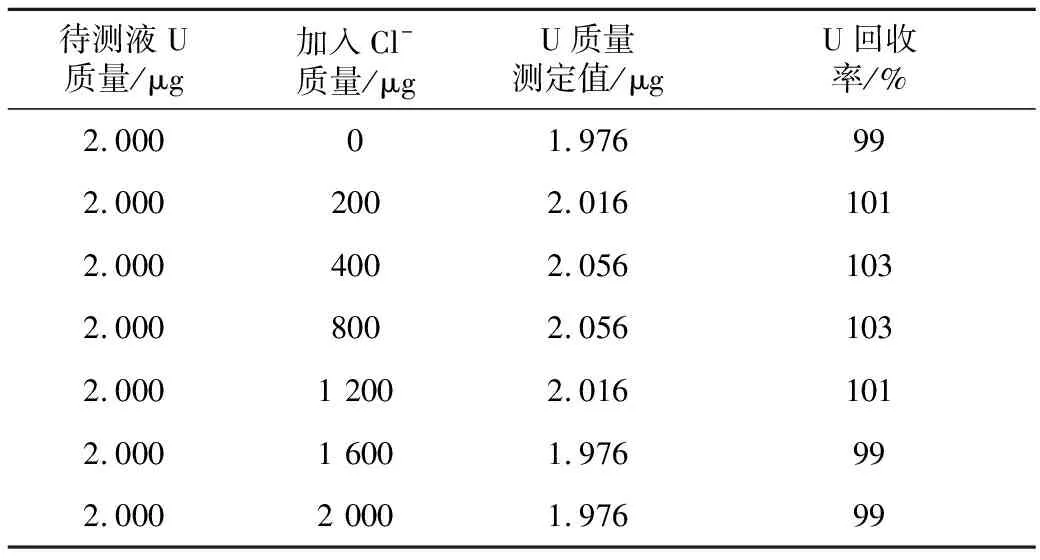
表5 Cl-对铀的干扰试验结果
从表5可看出,当样品中Cl-加入质量在200~2 000 μg时,即加入氯离子质量浓度在80 μg/mL的范围内,U回收率在99%~103%,不干扰U的分析测定。
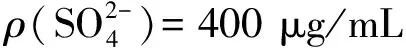
表对铀的干扰试验结果


表对铀的干扰试验结果
4溴代比色法优化
4.1 改变取样体积
4.2 标准曲线的校正与应用
4.2.1标准曲线的校正
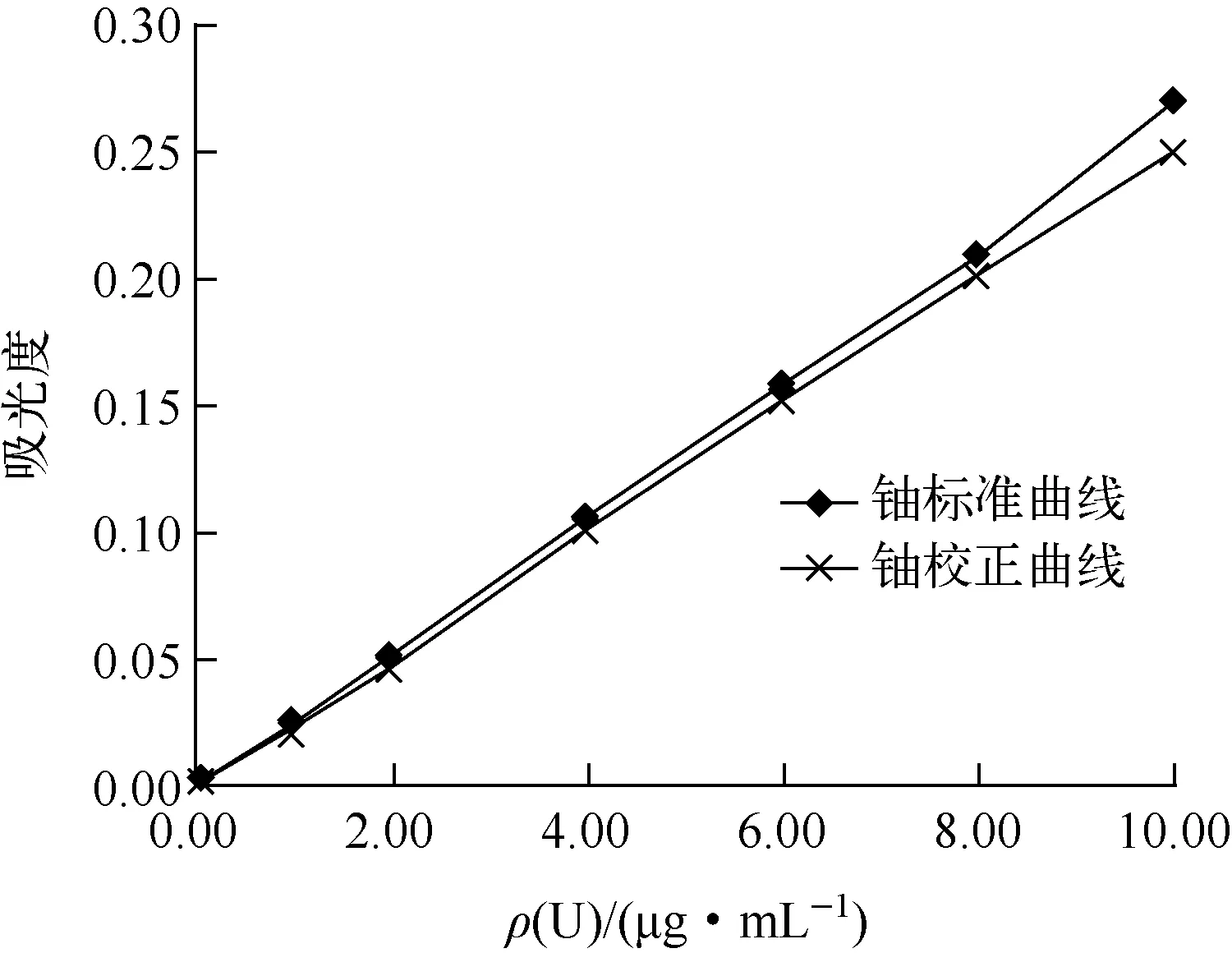
图3 铀标准溶液含的校准曲线
4.2.2校正工作曲线的应用
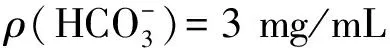
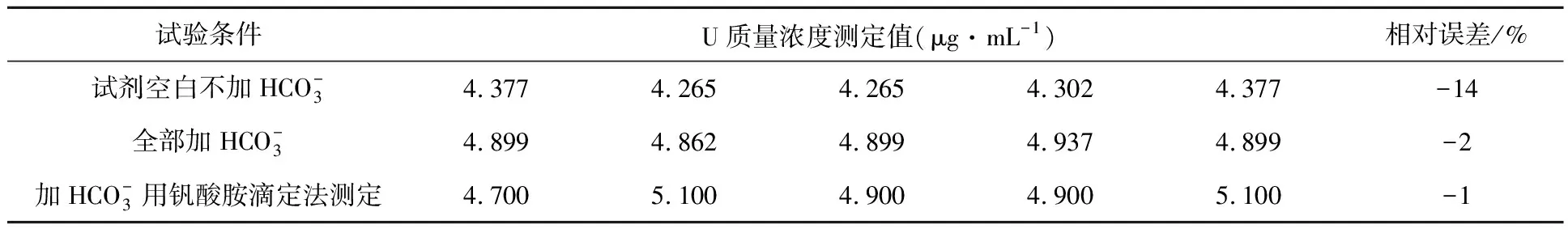
表8 对比分析试验结果
5结论
1)溴代比色法分析铀,测定下限是1.000 μg/mL,当样品中铀质量浓度低于1.000 μg/mL时,分析误差较大,其标准偏差和离散程度差,最大偏差达31%。
2)当检测出样品中铀质量浓度小于0.500 μg/mL时,测定值只有定性分析的意义,用于定量分析测定值误差很大。
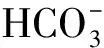
4)当取样体积大于1.0 mL时,分析结果相对误差开始增大,取样体积由10.0 mL改为1.0 mL时,相对误差由-65%降为2.4%。
