碳化硅表面结构对光催化活性的影响
2019-05-08冯纪章陈君华周永生叶祥桔徐卫兵
郭 雨,冯纪章,郭 腾,陈君华,柏 雷,周永生,叶祥桔,徐卫兵
(1.安徽科技学院 化学与材料工程学院,安徽 蚌埠 233100;2.合肥工业大学 化学工程学院,安徽 合肥 230009;3.安徽德力日用玻璃股份有限公司, 博士后工作站, 安徽 凤阳 233121;4.赛鼎工程有限公司,山西 太原 030032)
0 引 言

碳化硅(SiC)材料属于第三代宽禁带半导体材料,具有临界击穿电场和热导率高,介电常数小和电子饱和迁移率高,以及抗辐射能力强,机械性能好等特性,在电子器件、光电子器件[14]、储氢[15-16]、光催化[17-18]和传感[19]等领域都有广泛的应用前景.SiC属于共价键晶体,水溶液中表面Si—C共价键键不能被水直接破坏.因此,通过SiC表面羟基化能够很好验证表面端羟基Si—OH和C—OH对光催化性能的影响.基于此,本文采用氢氧化钠对碳化硅进行简单的化学浸蚀处理,以期解构表面Si—C键形成端羟基结构,并采用光电协同催化氧化亚甲基蓝实验验证其对光催化过程的影响.
1 实验部分
1.1 试剂和设备
超细碳化硅和氢氧化钠均购自国药试剂化学试剂有限公司,去离子水自制.
KQ2200ES超声波清洗机,昆山市超声仪器有限公司;MYP11-2A恒温磁力搅拌器,上海梅颖浦仪器仪表制造有限公司;DHG-9030智能鼓风干燥箱,杭州中拓仪器有限公司;AF500W光催化装置,上海季光特种照明电器厂;KXN-305D直流稳压电源,深圳市兆信电子仪器设备有限公司;UV-1800紫外可见光分光光度计.
1.2 表面浸蚀SiC的制备
一定浓度100 mL氢氧化钠溶液中加入5.00 g SiC粉末,连续恒速搅拌数小时.其中,氢氧化钠溶液浓度分别为0, 0.5, 1, 2和3 mol·L-1,浸蚀时间分别为4, 6, 8, 10和 15 h.浸蚀处理后,SiC样品反复洗涤除去氢氧化钠,干燥即得改性SiC.
1.3 样品表征
采用D/Max-Ra型X射线衍射仪(XRD)测定浸蚀前后SiC样品的晶体结构,激发源使用铜靶(Kα=0.154 06 nm),工作电压40 kV,电流30 mA.
浸蚀前后SiC粉体颗粒的形貌结构、表面晶格与粒径大小采用美国 FEI 公司生产的F20ST型场发射扫描电子显微镜进行测试.
浸蚀前后SiC样品的紫外-可见光漫反射吸收光谱用日本岛津UV-3600型紫外-可见光分光光度计测定,以标准BaSO4作为参比.
采用英国VG公司ESCALB MK-II 型X射线光电子能谱仪(XPS)进行浸蚀前后SiC样品表面元素的化学态分析,Al Kα为激发源,以样品表面污染碳的C 1s 结合能(284.7 eV)作能量内标.
采用Nicolet-380型傅里叶红外光谱仪分析浸蚀前后SiC样品表面的化学结构及其变化,波数范围400~4 000 cm-1.
1.4 光催化性能评价
光催化性能评价以MB为模型污染物,且使用之前未调节溶液pH值,整个反应均在常压室温下完成.将0.1 g碳化硅样品加到100 mL亚甲基蓝溶液中超声处理20 min,使得颗粒解聚分散形成均匀的悬浮液,并避光搅拌30 min,建立颗粒表面染料的吸附-脱附平衡.随后,将亚甲基蓝溶液置于不锈钢容器内,不锈钢容器与直流电源一侧电源线相联接,另一侧电源线接地,形成断路状态.采用500 W卤素灯作为光源,连续照射下降解污染物溶液.含催化剂的亚甲基蓝溶液每隔一定时间取10 mL,离心分离后,采用 UV-1800 紫外可见光分光光度计每隔一定时间测量分离催化剂后亚甲基蓝溶液的吸光度,测量的最大吸收波长为664 nm.
一定浓度范围内,亚甲基蓝溶液浓度和吸光度之间遵循朗伯-比尔定律,故可用吸光度变化替代浓度变化计算亚甲基蓝的降解率来衡量其光化学分解程度.因此,亚甲基蓝的降解率和拟一级动力学方程可以用式(1)和(2)计算.
η% =(Aeq-At) /Aeq×100,
(1)
ln(Aeq/At)=kappτ,
(2)
式中:Aeq,At分别表示催化剂吸附平衡后的亚甲基蓝溶液的吸光度和降解t时刻后溶液的吸光度;kapp为拟一级动力学的速率常数(min-1).
2 分析与讨论
2.1 浸蚀对SiC物相和形貌的影响
图 1 为SiC粉末浸蚀前后的XRD谱图.
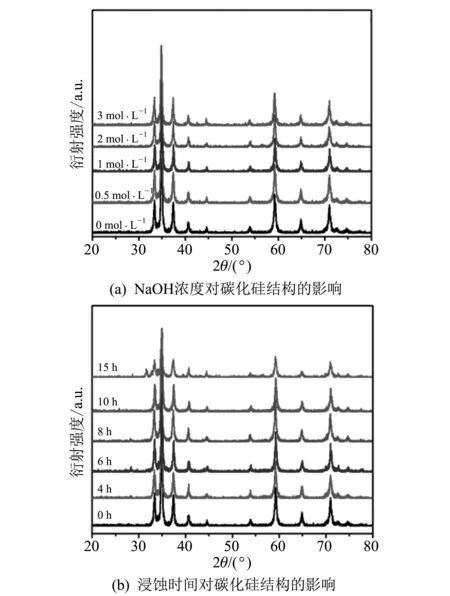
图 1不同浸蚀浓度和不同浸蚀时间条件下未改性和改性SiC样品的XRD谱图Fig.1 XRD patterns obtained for the unmodified and modified SiC under different etching concentrations and different etching times
如图 1 所示,所有SiC样品都能观察到 33.4°, 34.9°, 37.4°, 40.7°, 59.2°, 64.9°, 71°和75.0°处α-SiC (Moissanite-6H,JCPDS Card No.29-1128)的衍射峰,表明氢氧化钠表面浸蚀处理并没有改变SiC的晶体结构[20-21].此外,图 2 中浸蚀前后碳化硅样品无规则的颗粒形貌也没有发生明显改变,表明氢氧化钠没有对颗粒形成严重的腐蚀.

图 2浸蚀前后SiC样品的透射电镜图Fig.2 Typical TEM image of SiC particles before and after etching treatment
2.2 浸蚀对SiC表面结构的影响
FT-IR光谱用于碳化硅表面表面结构的分析,浸蚀前后SiC的红外光谱如图 3 所示.所有SiC样品的红外光谱均出现了1 630 cm-1和 3 438 cm-1吸收峰,对应于SiC表面H2O的弯曲振动,而841 cm-1处的吸收峰可以归为Si—C键的振动模式[22-24].1 122, 1 455, 1 540 和1 744 cm-1处的吸收峰均为碳氧物种的红外振动[21-22,25].更值得注意的是:① 480 cm-1处Si—C的吸收峰随着浸蚀浓度和浸蚀时间的增加逐渐消失,而对应的500 cm-1处Si—O—Si的吸收峰强度逐渐增加;② 1 454 cm-1和1 740 cm-1处与-C=O吸收峰也呈现出与480 cm-1处Si—C键相同的变化,而且浸蚀15后1 003 cm-1和1 455 cm-1附近有两个明显的吸收峰,可能是表面过度腐蚀形成的-COOH 基团和-C-OH基团;③ 3 400 cm-1和 1 630 cm-1处与物理吸附水相关的红外吸收峰强度也随着浓度和时间的增大而呈现递减趋势.这表明SiC表面的原有的氧化层和Si—C层逐渐被氢氧化钠解构形成氧化硅层,同时碳可能以碳酸盐的形式脱除[26-27].

图 3不同浸蚀浓度和浸蚀时间下SiC样品的红外光谱Fig.3 FT-IR analysis for unmodified and modified SiC under different etching concentration and different etching times
图 4 和图 5 比较了不同浸蚀条件下SiC中C,O和Si三种元素的结合能峰值变化.

图 4不同NaOH浓度处理后SiC的Si 2p, C 1s和O 1s XPS谱图Fig.4 Si 2p, C 1s and O 1s core-level spectra of unmodified and modified SiC under different NaOH concentrations
由于XPS峰位与元素的电负性有关,电负性大的氧元素对应的峰值也越大.很显然,三种元素的化学位移均呈现出相同的下降趋势,其原因是表面氧化物层减薄所致[28-31].因此,结合红外分析的结果可知,碳化硅最外层表面上氧化层和部分Si—C结构消失,形成了新的Si—OH层.

图 5不同浸蚀时间后SiC的Si 2p, C 1s和O 1s XPS谱图Fig.5 Si 2p, C 1s and O 1s core-level spectra of SiC under different etching times
2.3 浸蚀条件对SiC光催化性能的影响
浸蚀浓度和浸蚀时间是影响碳化硅表面羟基化的两个重要因素.不同浸蚀浓度和浸蚀时间对MB降解率的影响如图 6 所示.从图 6(a) 可以看出,当浸蚀浓度为1 mol·L-1时,MB的紫外光降解率达到了45.9%,明显高于未处理SiC样品和其他改性样品.随着浸蚀浓度的增加,MB降解率明显减小.对于改性SiC样品,施加正、负偏压均使得MB降解趋势明显增加,最大降解率分别达到59.78%和62.91%.浸蚀时间的影响也呈现出相似的降解趋势.从图 6(b) 中可以看出,相同浓度的氢氧化钠溶液中,不同浸蚀时间的SiC样品对MB降解率呈现出先增后减的趋势.其中,浸蚀10 h后SiC样品的MB降解率达到最大值.结果表明,氢氧化钠浓度1 mol·L-1和浸蚀时间10 h是最佳的表面处理条件.

图 6不同浸蚀条件处理后的碳化硅紫外光催化性能比较Fig.6 Comparison of catalysis over different SiC samples
2.4 偏置电压对MB降解率的影响
图 7 为施加0, ±220, ±380, ±500, ±1 000, ±2 000, ±4 000 V, ±8 000, ±10 000和±20 000偏压电场对MB降解率的影响.实验结果表明,正偏压或负偏压增加都会导致MB降解率呈现出先增大后减小的变化趋势,特别是高压下MB降解率减小更加显著.当加载电压±500 V时,MB降解率达到最大,其40 min的降解率将近80%,比没有加载电压的情况高出约55%.对于胶体粒子,高压静电场中胶体粒子的聚沉导致了催化剂颗粒分布不均.而SiC颗粒并非是胶体粒子,没有搅拌则极易沉积于容器底部,施加电场并没有对其分散行为产生显著影响.因此,施加偏置电压导致SiC表面能带弯曲,使得带隙减小,从而加剧了电子和空穴的复合,使得光生载流子电致分离效应被抵消,从而造成了MB降解率的减小[32-34].

图 7偏置电压对MB降解率的影响Fig.7 Effect of bias voltage on MB degradation rate
2.5 浸蚀SiC与未浸蚀SiC光催化性能的对比
为了对比氢氧化钠处理前后SiC光催化性能的改变,表面处理SiC与表面未处理SiC的紫外光催化以及光电催化实验被用于验证光催化性能的差异.SiC紫外光催化以及光电催化MB降解变化曲线如图 8 所示.从图 8(a) 中可以看出,未表面浸蚀处理的SiC样品通过光催化和光电催化过程的去除率分别为1.9%, 3.7%和2.2%,光催化降解MB基本可以忽略.相反,表面氧化层薄化后的SiC样品的MB光催化效率则分别达到了45.9%, 78.75%和80%,表明SiC表面结构调控对光催化效率产生了决定性的影响.
图 8(b) 中动力学研究表明,MB的光催化和光电催化反应遵循拟一级动力学模型,其拟一级动力曲线如图所示.经过线性拟合,未表面浸蚀处理的SiC样品的光催化和光电催化过程的动力学常数分别为2.58×10-4, 5.03×10-4和2.96×10-4min-1,而表面处理后的SiC样品的MB降解动力学常数分别是79.0×10-4, 363.9×10-4和355.9×10-4min-1,是光催化过程动力学常数的30.62, 72.35和120.24倍.其原因为未表面浸蚀处理的SiC样品表面存在较厚的碳、硅氧化层,被激发的光生电子即使在外加偏压的作用下也很难到达表面参与反应.而SiC表面氧化层减薄处理有利于光生电荷向表面迁移,外加电场也抑制了光生电子和空穴的复合,从而提高了光电催化降解速率.

图 8表面处理前后SiC样品的光催化和光电催化性能Fig.8 Photocatalytic and photoelectrocatalytic performance ofSiC and modified SiC
3 结 论
1) 采用简单的化学浸蚀法,控制浸蚀液浓度与浸蚀时间实现了SiC表面的氧化层的减薄,最佳的浸蚀条件为氢氧化钠浓度1 mol·L-1和浸蚀时间10 h;
2) 浸蚀处理后的SiC样品晶体结构和形貌均没有改变,其物相均为α-SiC.XPS和FT-IR的结果分析表明,SiC表面的碳、硅氧化层被腐蚀减薄;
3) 表面处理后的SiC表现出了优异的光催化和光电化学性能,光催化过程动力学常数分别是未处理样品的30.62,72.35和120.24倍,证明了SiC的表面结构是增强光催化效率关键因素.
