高分辨质谱快速筛查牛乳中10种禁用兽药残留
2019-04-26贡松松张婧严凤吴剑平潘娟卢洪秀
贡松松,张婧,严凤,吴剑平,潘娟,卢洪秀
(1.上海市兽药饲料检测所,上海201103;2.上海农林职业技术学院农业生物与生态技术系,上海201600)
0 引 言
由于现代农业中兽药的大规模使用,牛乳中抗生素和激素的残留受到广泛关注。欧盟对兽药残留限量作了严格规定[1]。我国为加强兽药残留监控,也对此作了严格规定[2]。有研究表明,儿童性早熟,男性前列腺癌,妇女乳腺癌和子宫癌发病率的上升与食品中雌激素(Estrogens)的残留有关[3-5]。氯霉素类(Chloramphenicols)药物通过食物链进入人体,则会对人体造血机能产生严重的副作用[6-7]。
牛乳中雌激素和氯霉素常用的检测方法有气相色谱-质谱联用法(GC-MS)[8]和液相色谱-质谱联用法(LC-MS)[9-10]。GC-MS虽然分离效果好,但是雌激素需要衍生化,操作繁琐。以四级杆-飞行时间质谱为代表的高分辨质谱既可以获得分子离子信息,又能获得丰富的碎片离子信息。因此,其成为多残留的有效分析方法[11-12]。本文借鉴QuEChERS[13-16]方法在多残留前处理方面的优势,建立了生鲜牛乳中10种禁用兽药的快速筛查方法。
1 实 验
1.1 材料与试剂
乙腈(色谱纯),甲酸(色谱纯),乙酸(分析纯),无水硫酸钠,氯化钠(分析纯),乙二胺-N-丙基硅烷(PSA),十八烷基硅烷(ODS C18),超滤离心管(3ku),实验用水为超纯水。
氯霉素标准品(Chloroamphenicol,CAP),甲砜霉素标准品(Thiamphenicol,TAP),氟苯尼考标准品(Florfenicol,FF),雌酮标准品(Estrone,E1),己烷雌酚标准品(Hexestrol,HES),炔雌醇标准品(Ethinylestradiol,EE2),雌三醇标准品(Estriol,E3),己烯雌酚标准品(Diethylstilbestrol,DES),雌二醇标准品(Estradiol,E2),双烯雌酚标准品(Dienestrol,DIE),纯度均大于96.1%。
1.2 仪器与设备
超高效液相色谱—四级杆飞行时间质谱(6530),包括G4220A流动相输送泵,G4226A自动进样器,G1316C柱温箱,G4212A二极管阵列检测器;Mass-Hunter数据处理系统,AE240电子天平,Allegra X-22R高速冷冻离心机,多管漩涡振荡器,Milli-Q超纯水系统。
1.3 标准溶液的配制
标准品贮备液:称取10 mg标准品(根据纯度折算质量),用色谱纯甲醇溶解并定容至10 mL,制成质量浓度为1mg/mL的单标贮备液。对于每类化合物,将单标溶液混合制成50或质量浓度为100 μg/mL的混合标准溶液,使用时再根据需要混合或稀释,这些混合标准溶液用来制备标准曲线和质量控制样品。将配制好的标准品贮备液置于4℃环境下低温避光保存。
1.4 样品前处理
准确称取5.0 g牛乳样品于50 mL离心管中,加入20 mL乙腈,涡旋振荡1 min,加入4.0 g无水硫酸钠和1.0 g氯化钠,涡旋震荡4 min,转速为8 000 r/min离心5 min。准确移取10 mL上层清液于离心管中,加入1.0 g无水硫酸钠,0.2 g PSA,涡旋振荡器振荡4 min,转速为8 000 r/min离心5 min,取5 mL上清液于40℃氮气吹至近干,用1.0 mL乙腈-水溶液(5∶95,体积比)定容,涡旋30 s,过0.22 μm滤膜后,供超高效液相色谱—四级杆飞行时间质谱测定。
1.5 超高效液相色谱-四级杆飞行时间质谱条件
1.5.1 超高效液相色谱条件
色谱柱:Agilent ZORBAX SB-C18柱(2.1 mm×100 mm,1.8 μm);流动相A为水,B为乙腈。梯度洗脱程序:0 min,5%B;2 min,5%B,2~4 min,5%~40%B;4~9 min,40%~80%B;9~9.5 min,80%~95%B;9.5~15 min,95%~95%B;15~17 min,95%~5%B;17~20 min,5%B;流速为0.3 mL/min;柱温为40 ℃;进样量为10 μL。
1.5.2 质谱条件
离子源为Dual AJS ESI,负离子模式;扫描范围为50~1000 m/z;干燥气温度为320℃;干燥气流速为8 L/min;雾化器压力为0.24 MPa;鞘气温度为400℃;鞘气流速为12 L/min;毛细管电压为3 000 V;喷嘴电压为500 V;碎裂电压为130 V;飞行时间管真空度为2.04×10-7;仪器在高分辨率(4 GHz),低质量范围(<m/z 1700)下操作。10种雌激素类和氯霉素类药物的质谱分析参数和提取离子色谱图分别如表1所示。
1.6 筛查谱库的建立
取合适浓度的混合标准品工作液稀释到质量浓度为1 mg/L,在优化好的色谱质谱条件下进样,先进行一级质谱扫描,得到目标药物的保留时间、质子化的分子质量数、质量误差等信息。在不同的碰撞电压下再进行二级质谱扫描,得到目标化合物的二级全扫描图及质谱碎片信息,以此建立10种雌激素类和氯霉素类药物的筛查谱库。
2 结果与讨论
2.1 色谱条件的优化
2.1.1 色谱柱的选择
实验比较了SB C18(3.0 mm ×100 mm,1.8 μ m)、SB C18(2.1 mm × 100 mm,1.8 μm)和Extend C18(2.1 mm × 50 mm,1.8 μm)3种色谱柱对10种雌激素类和氯霉素类药物的分离度和检测灵敏度的不同影响。实验表明,ZORBAX SB C18(2.1 mm×100 mm,1.8 μm)色谱柱更适合分析目标药物,内径越小,峰形越尖锐对称。虽然5 cm色谱柱用时更短,但不能保证每种药物都有良好的峰形,在粒径相同的情况下,采用内径更小的10 cm色谱柱,结合高分辨质谱的优异性能,能获得良好的分离度。
2.1.2 流动相的选择
本实验考察不同pH的水系流动相(水、0.1%甲酸水溶液、1%甲酸水溶液、0.1%乙酸水溶液、1%乙酸水溶液)对目标药物在色谱柱上分离效果的影响。结果表明,流动相加入甲酸或乙酸后,峰形较差。当使用超纯水时,目标药物的离子化效率较好,分离效果令人满意。故选择乙腈和水作为流动相,采用梯度洗脱方式可以很好地将10种雌激素类和氯霉素类药物分离,具有较好的灵敏度和分辨率。
2.1.3 梯度洗脱条件的选择
由于10种雌激素类和氯霉素类的极性差异较大,采用恒定的流动相很难将各个药物分开,故采用梯度洗脱的方式,改变流动相的极性,从而达到使目标药物完全分开的目的。实验比较了不同比例的初始流动相中的有机相(5%,10%,20%)和不同的洗脱时间(12,15,20 min)对洗脱效果的影响。随着乙腈比例的升高,多数药物被快速的洗脱下来,没有达到良好的分离,当乙腈的初始比例为5%时,目标药物能被缓慢地洗脱下来,分离良好。随着洗脱时间的增加,全部药物能有较好的灵敏度和分辨率。故选择初始流动相有机相比例为5%,洗脱时间20 min。
2.1.4 定容液的选择

表1 10种雌激素类和氯霉素类药物信息
样品定容液不仅会影响目标药物的分离效果,而且还会影响离子化效率。本实验比较了超纯水、乙腈-水(5∶95,体积比)、乙腈-水(20∶80,体积比)、乙腈-水(50∶50,体积比)和乙腈-水(90∶10,体积比)作为定容液时的情况,实验发现,当定容液有机相比例超过初始流动相中的有机相比例时,多数药物的峰形变宽。当有机相比例不大于初始流动相时,峰形较尖锐,本实验选择乙腈-水(5∶95,体积比)作为定容液,跟初始流动相比例相同。
2.2 质谱条件的优化
Dual AJS ESI容负离子模式下,在50~1 000 m/z范围内作一级质谱全扫描,得到10种雌激素类和氯霉素类药物的总离子色谱图。比较各个化合物的提取离子色谱图的响应和峰形,依次优化鞘气温度和流速、喷嘴电压、毛细管电压、雾化器压力和干燥气温度和流速。然后优化不同的碎裂电压,在定性分析软件中,选择最优电压参数。详细参数见1.5.2。
2.3 样品前处理方法的选择
2.3.1 超滤膜方法的优化
称取5.0 g牛乳样品于50 mL离心管中,加入20 mL乙腈,涡旋振荡30 s,转速为8 000 r/min离心5 min。取5 mL上清液于40℃氮气吹至近干,用1.0 mL乙腈-水溶液(5∶95,体积比)定容,转移至3 ku的超滤离心管中,14 000 r/min离心60 min,净化过后的提取液用于进样分析。实验比较了未经初始流动相溶液润洗超滤膜(A组)和经初始流动相溶液润洗(B组)对超滤离心管净化效果的影响。实验结果如图1所示,预洗步骤对目标药物的回收率没有产生明显影响。另外,雌二醇、炔雌醇、雌酮、己烯雌酚、双烯雌酚、己烷雌酚的回收率均低于50%。
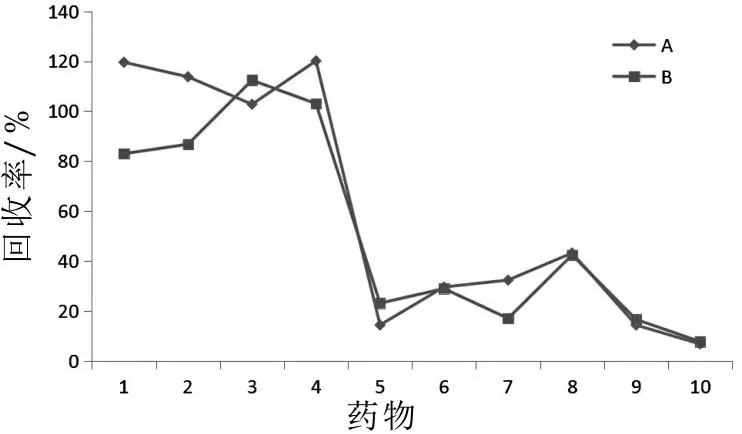
图1 预洗对10种雌激素类和氯霉素类药物回收率的影响
2.3.2 QuEChERS提取溶剂的优化
为了同时提取10种雌激素类和氯霉素类药物,实验比较了不同酸度的乙腈作为提取液,分别为:乙腈(A)、0.1%甲酸乙腈(B)、1%甲酸乙腈(C)、0.1%乙酸乙腈(D)、1%乙酸乙腈(E)。10种雌激素类和氯霉素类药物的回收率实验结果如图2所示。由图2可以看出:B,C,D,E组的双烯雌酚和己烷雌酚回收率不足20%;C,D,E组的己烯雌酚已检测不出;A组药物总体回收率介于50%~120%之间,故选择A组(乙腈)作为提取溶剂。
2.3.3 QuEChERS净化步骤的优化
传统的QuEChERS方法使用PSA作为吸附剂去除果蔬中脂肪酸色素等物质,而牛乳基质更为复杂,含有大量的脂肪和蛋白,脂肪和蛋白质的干扰对牛乳样品至关重要。PSA是在高纯硅胶基质上键合N-丙基乙二胺的极性吸附剂。C18是一种非极性吸附剂,可吸附非极性到中等级性的化合物。PSA和C18都可用于除去脂肪酸、色素和糖类等基质成分,这些分散固相萃取剂在净化样品的同时有可能对目标产物产生一定的吸附作用,因此有必要对PSA(A组)和C18(B组)进行优化。以药物的回收率作为评定依据,实验结果如图3所示,由图3可以看出:B组己烯雌酚、双烯雌酚和己烷雌酚回收率不足30%,与B组数据相比,A组数据回收率介于50%~120%之间,故优化条件选择A组(PSA)作为吸附剂。
2.3.4 超滤膜与QuEChERS方法的比较
从图1和图3可以发现,超滤膜虽然能够节省时间,简化步骤,但是有超过50%的药物回收率低于50%。QuEChERS方法药物回收率介于50%和120%之间,适用于药物多残留分析,故采用QuEChERS方法。
2.4 方法学验证
2.4.1 标准曲线与线性范围
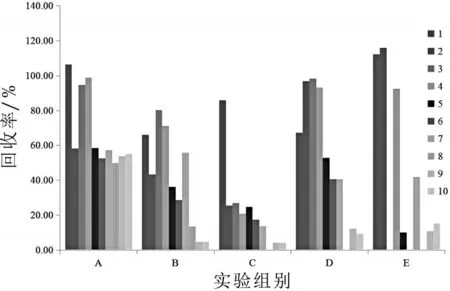
图2 提取溶剂对10种雌激素类和氯霉素类药物回收率的影响

图3 优化实验条件的回收率实验结果
采用液相色谱-串联质谱方法的弊端之一是基质效应现象,因此有必要对基质效应进行考察。取空白生鲜牛乳样品基质溶液与流动相分别配制标准溶液,以分子离子峰面积Y对质量浓度X(μg·L-1)作标准曲线,获得目标化合物的基质匹配标准曲线和溶剂标准曲线。利用公式ME=k1/k2×100%计算基质效应(见表2)。其中k1和k2分别为基质匹配标准曲线的斜率和溶剂标准曲线的斜率。ME偏离100%越多,说明基质效应越明显。如表2所示,氟苯尼考和雌三醇出现了
基质增强效应,己烯雌酚、双烯雌酚和己烷雌酚出现较强的基质抑制,其余的化合物基质效应不明显。因此本实验采用基质匹配标准曲线外标法定量。10种雌激素类和氯霉素类的线性方程、线性范围和相关系数如表2所示。
2.4.2 检出限(LOD)与定量限(LOQ)
用空白牛乳基质提取液稀释标准曲线的最低浓度,以3倍仪器检测信噪比(S/N=3)计算各化合物的定性筛查检出限(LOD),结果表明,10种雌激素类和氯霉素类的LOD为5~20 μg/kg。以10倍信噪比(S/N=10)估算定量下限,10种雌激素类和氯霉素类的LOQ为10~50 μg/kg。实验结果如表2所示。
2.4.3 回收率与精密度
采用空白生鲜牛乳样品进行加标回收率及精密度实验,样品分别添加低、中、高3个质量浓度的标准溶液,每个浓度平行测定6次,计算平均回收率及相对标准偏差(RSD),测定结果如表3所示。由表3可以看出,10种雌激素类和氯霉素药物的回收率为61.2%~116.3%,相对标准偏差为2.3%~10.8%。
2.5 实际样品的筛查
应用本方法对全国8个省市的80份牛乳样品,采用已建立的超高效液相色谱-四级杆飞行时间质谱筛查方法,再经二级质谱确证,结果发现:雌酮和雌三醇均表现出一定的个体差异性,雌酮的平均值为132.0 μg/kg,最大值达到了393.9 μg/kg。雌三醇的均值质量分数为52.0 μg/kg,最大值达到了92.5 μg/kg。

表2 10种雌激素类和氯霉素类药物的实验结果的基质效应、线性方程、线性范围、相关系数、检出限及定量限
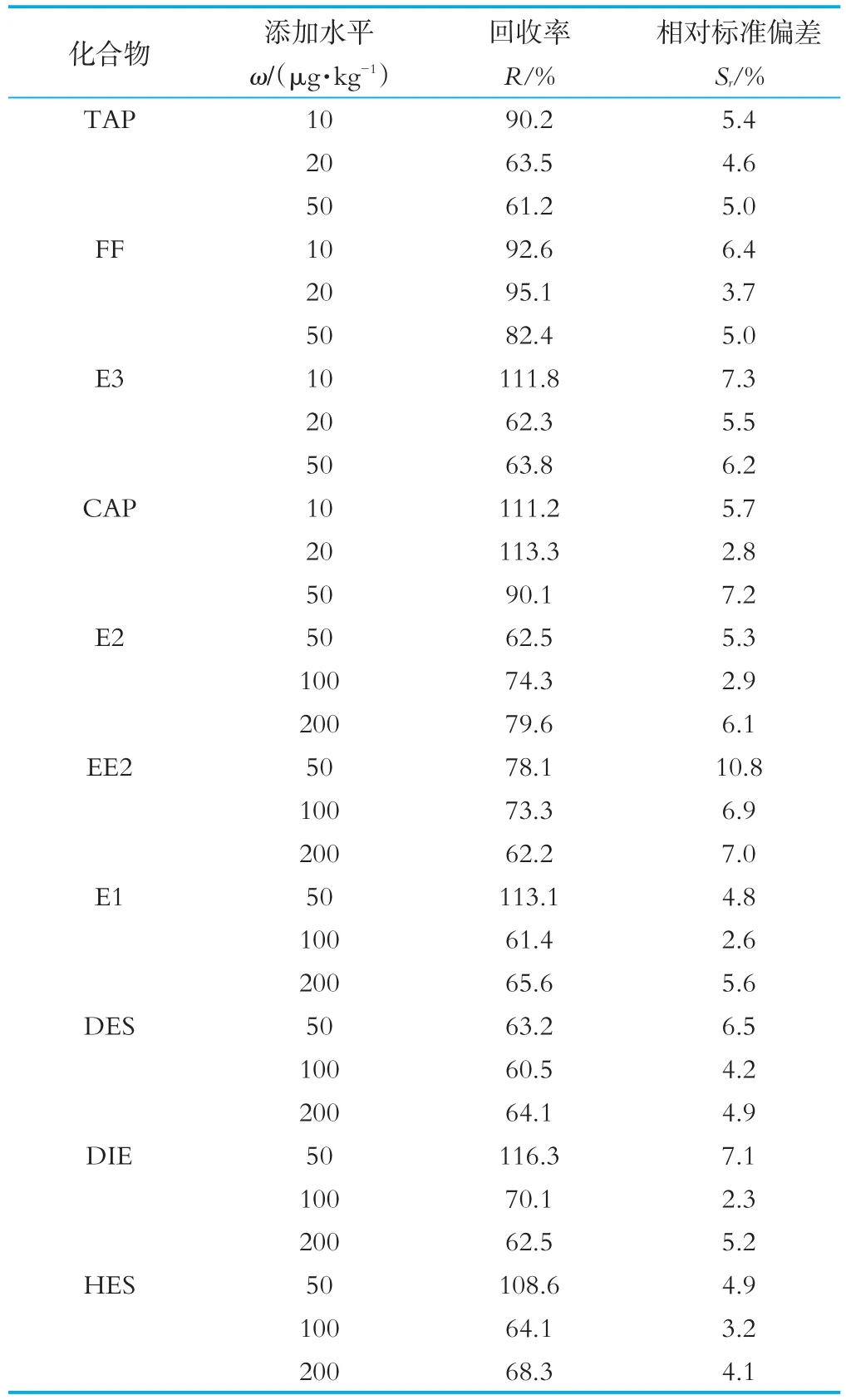
表3 10种雌激素类和氯霉素类药物的加标回收率及相对标准偏差(n=6)
雌三醇、雌酮为天然激素,可能为奶牛体内的内源性激素。作为一种筛查方法,适用于生鲜牛乳中雌激素类和氯霉素类药物的快速定性确证,可为监管和监督提供技术依据。
3 结论
利用超高效液相色谱的快速分析与飞行时间质谱的高分辨率优势,建立了生鲜牛乳中10种雌激素类和氯霉素类药物的超高效液相色谱—四级杆飞行时间质谱筛查与确证方法,改进了前处理方法,优化了色谱质谱条件。本方法快速简便,前处理成本低廉。作为一种筛查方法,适用于生鲜牛乳中10种雌激素类和氯霉素类药物的快速定性确证。
