紫茎泽兰不同入侵区域土壤细菌群落多样性比较研究
2019-04-13孔令杰韩月龙张风娟
柳 旭, 孔令杰, 杨 康, 韩月龙, 张风娟
河北大学生命科学学院, 河北 保定 071002
紫茎泽兰Ageratinaadenophorum(Sprengel) R. K. H.为多年生半灌木,属菊科泽兰属(Sangetal.,2010),原产于美洲墨西哥至哥斯达黎加一带(王文琪,2006),于20世纪40年代由缅甸传入我国云南省,现广泛分布于我国西南地区,并有进一步扩张的趋势(Wang & Wang,2010)。紫茎泽兰具有强大的生态适应性和竞争优势,对当地的农牧业发展、人畜健康以及生态环境造成严重威胁(Luetal.,2005)。
土壤微生物在外来植物入侵过程中起着重要作用(肖博,2014)。入侵植物与土壤微生物之间的相互作用关系深刻影响着入侵植物的适应性和竞争力(付伟等,2017; Zhangetal.,2017,2018)。土壤微生物参与土壤中有机质的分解、土壤腐殖质形成及分解以及养分的转化和循环,是生态系统的重要组成部分(滕应和黄昌勇,2002; Geetal.,2010; Mcguireetal.,2010)。入侵植物在入侵地形成稳定种群后会影响入侵地的植物群落结构,进而引起土壤微生物的多样性发生变化(Levineetal.,2003)。如藿香蓟AgeratumconyzoidesL.对梨树各发育时期和各土层土壤微生物数量和比例的影响呈现多态效应,对土壤中细菌数量的提高效应优于对土壤真菌和放线菌(吴红英等,2010);空心莲子草Alternantheraphiloxeroides(Mart.) Griseb.入侵后,土壤中可培养细菌和真菌的数量显著增加,而放线菌的数量显著下降(王志勇等,2011);假高粱Sorghumhalepense(L.) Pers.的根系分泌物能选择性地影响其根际细菌的数量和群落结构,形成假高粱特有的根际土壤细菌群落结构和多样性,一些土壤细菌的介入会提高假高粱的竞争优势(刘纯等,2013)。因此,土壤细菌群落与植物的入侵密不可分(牛红榜等,2007a;于兴军等,2005),探究入侵植物根际土壤细菌群落变化对揭示其入侵机制具有重要意义。
关于紫茎泽兰入侵与土壤微生物之间的关系已有广泛研究(Yuetal.,2005)。紫茎泽兰入侵改变了土壤微生物群落结构,提高土壤自生固氮菌、氨氧化细菌和真菌的数量、提高土壤可利用的养分水平,创造对自身生长有利的土壤环境(牛红榜等,2007a);刘潮等(2018)和Yuetal. (2014)证明紫茎泽兰根围的土壤微生物增强了其对本地植物种的竞争力,形成了自我促进的入侵机制;紫茎泽兰叶水溶液中的次生化感物质对土壤细菌群落结构影响也较大(Zhuetal.,2017),其入侵改变了根际土壤细菌多样性,可能通过聚集一些特定的菌群来实现成功入侵。本研究采集云南境内不同入侵域紫茎泽兰根际土壤,比较不同生境中紫茎泽兰根际土壤细菌群落结构,探究土壤细菌群落结构与环境因子间的关系。研究结果对于揭示紫茎泽兰的入侵机制具有重要作用,同时也大大丰富入侵植物的土壤微生物假说,并为紫茎泽兰入侵的控制和管理以及生态系统的修复提供理论依据。
1 材料与方法
1.1 供试材料采集
紫茎泽兰样地位于中国西南地区的云南省(97°31′-206°11′E,21°8′-29°15′N)。该地土壤类型多为红壤,气候属于亚热带高原季风型,干湿季节分明,紫茎泽兰入侵严重,形成明显的单优群落。于2017年3月在云南昆明、玉溪、普洱采用“五点法”分别采集紫茎泽兰根际土壤样品,设置样方大小为3 m×3 m,每个样点选取3个样方。去除样品中的石块、断根及其他杂质,在实验室将土壤过2 mm筛。土样过筛后分成3份:一份于室温保存,用于土壤理化性质测定;一份置于-20 ℃保存,用于PLFAs分析;一份置于-80 ℃保存,用于分子分析。采集的土壤样品的基本信息见表1。

表1 取样地点概况Table 1 The general situation of sample location
1.2 土壤样品理化性质的测定
土壤pH值用电位法测定(水土比=2.5∶1);土壤蛋白酶活性用茚三酮比色法测定,以24 h后1 g土壤中氨基氮的mg数表示;脲酶活性用苯酚钠—次氯酸钠比色法测定,以1 g土壤中NH3-N的mg数表示;磷酸酶活性用磷酸苯二钠比色法测定,以1 g土样1 h催化PNPP分解生成对硝基苯酚的μg数表示;蔗糖酶活性用3,5-二硝基水杨酸比色法测定,以24 h后1 g土中葡萄糖mg数表示(关松荫,1986; 哈兹耶夫等,1980);土壤速效磷用碳酸氢钠浸提—钼锑抗比色法测定;速效钾用1 mol·L-1中性醋酸铵浸提—火焰光度计法测定;有机质采用重铬酸钾法测定;铵态氮和硝态氮用2 mol·L-1KCl溶液浸提土样后,采用全自动化学分析仪(Smart-Chem 200, Alliance, France)测定(南京农业大学,1990)。
1.3 磷脂脂肪酸(phospholipid fatty acid ,PLFAs)分析
参考Frostegårdetal.(1993)和Kourtevetal.(2002)方法对PLFA进行提取和分析。PLFA成分分析采用Sherlock MIS 4.5微生物自动鉴定系统(sherlock microbial identification system,美国MIDI公司),其中PLFA定量采用19∶0内标法。根据结构不同,不同特征脂肪酸对应不同的微生物(吴愉萍,2009; Frostegård & Baath,1996; Hilletal.,2000; Huangetal.,2013; Olsson & Alstrom,2000)。
1.4 土壤DNA提取及PCR扩增
采用手提法(张海燕等,2009; Zhouetal.,1996)提取土壤样品总基因组DNA,利用1%琼脂糖凝胶电泳检测土壤总DNA的完整性,利用DNA浓度测定仪(Nanodrop)检测DNA浓度。使用16S rDNA-V4区特异引物515F(5′-GCGCCAGCMGCCGCGGTAA-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)进行PCR测定,用2%的琼脂糖凝胶电泳进行检测。
1.5 Miseq测序
本研究采用Miseq (Illumina,美国)进行DNA测序,将测序后的DNA序列拼接,同时对序列的质量和拼接效果进行质控过滤,然后按照barcode标签序列识别并区分样品,得到有效数据。采用Usearch软件设置97%相似性,对有效DNA序列数据进行操作分类单元(operational taxonomic unit,OTU)分类。Miseq测序、序列拼接以及OTU分类均由北京诺禾致源生物信息科技有限公司完成。
1.6 数据处理与分析
实验数据以平均值±标准差的形式表示;采用Microsoft Excel 2016软件统计数据;采用SPSS 21.0软件进行单因素方差分析(One-way ANOVA)和主成分分析(principal components analysis,PCA);采用Microsoft Excel 2016和Past 3软件作图。
2 结果与分析
2.1 不同样点紫茎泽兰根际土壤样品微生物群落结构
Y1、Y2、Y3和Y4样点紫茎泽兰根际土壤样品中分别检测到53、61、58和57种PLFA。选取大于0.01 nmoL·g-1的32种PLFA进行分析,其中代表革兰氏阳性细菌的PLFA 15种、革兰氏阴性细菌12种、真菌1种、放线菌3种、丛枝菌根真菌1种(表2)。分析结果表明,4个样点中各类微生物含量均存在显著差异(表3),其中Y2采样点的细菌、真菌、放线菌、丛枝菌根真菌及总磷脂脂肪酸含量均显著高于其他样点,而Y1样点的各类微生物含量均显著低于其他样点。各采样点的各类群微生物中以细菌PLFAs含量最高,占总PLFAs的63.55%~74.92%(图1),各生境土壤样品细菌磷脂脂肪酸含量均表现为与微生物总磷脂脂肪酸含量相同的变化趋势。
对紫茎泽兰根际土壤微生物群落PLFA进行主成分分析(图2),结果显示,4种样品分别位于四个不同的象限且空间距离较远,说明在不同样点土壤微生物的群落结构存在差异。采用SPSS21.0软件计算主成分因子载荷值,对PC1贡献较大的PLFA为16∶1 ω9c,17∶0,14∶0 iso,16∶0 iso和17∶0 anteiso;对PC2贡献较大的PLFA为12∶0和14∶0,根据表2,以上PLFA均为代表细菌的PLFA,说明细菌群落的差异是影响不同样点紫茎泽兰根际土壤微生物群落差异的主要因素。

表2 土壤微生物PLFA生物标记物Table 2 Phospholipid fatty acid (PLFA) signatures of soil micro-organisms

表3 同样地土壤微生物各菌群磷脂脂肪酸含量(n=3)Table 3 The contents of soil microbial PLFAs in different soil samples (n=3)
同列数据(平均值±标准误)后不同小写字母者表示在5%水平上差异显著。
The data (means±SD) in the same column with the different letters mean significant differences at 5% level.

图1 不同样点土壤微生物群落相对含量堆积图Fig.1 Soil microbial community relative content accumulationhistogram in different locationsY1:普洱市思茅区;Y2:玉溪市红塔区;Y3:昆明市官渡区;Y4:昆明市西山区。Y1: Simao District, Pu′er; Y2: Hongta District, Yuxi; Y3: Guandu District, Kunming; Y4: Xishan District, Kunming.
2.2 土壤细菌的OTU丰度和α多样性
对土壤样品DNA进行高通量测序,经过拼接和过滤处理后,获得16S rDNA标签序列,根据97%的序列相似性划分为不同的OTU。OTUs丰度稀释曲线(图3)显示,随着测序数量的上升,稀释曲线斜率逐渐下降,趋向平坦但未达平台期,说明测序数量足够,能够反映样品中的物种组成特征,但仍有小部分低丰度类群未被覆盖。
土壤细菌多样性指数和丰富度指数如表4所示。4个样点之间土壤细菌Shannon指数和Simpson指数无显著差异。Y2的Chao1指数显著低于Y1、Y3和Y4,而Y1、Y3和Y4无显著差异。PD_whole_tree指数代表通过进化关系观察到的种属数,本实验中Y2土壤细菌的种属数显著低于其他3个样地;Y3土壤细菌的种属数显著高于Y2,但低于Y1、Y4且差异显著;Y1、Y4土壤细菌种属数无显著差异。
2.3 土壤细菌OTUs分布
各土壤样品一共有12398个细菌OTUs(图4),其中共有的OTUs为2737个,只占总数的22.08%,说明不同地理条件对土壤细菌OTU影响明显。Y2与Y1、Y3、Y4的细菌OTU共有数少于其他3个样品之间的细菌OTU共有数,说明Y2区域与Y1、Y3和Y4区域细菌组成存在差异。
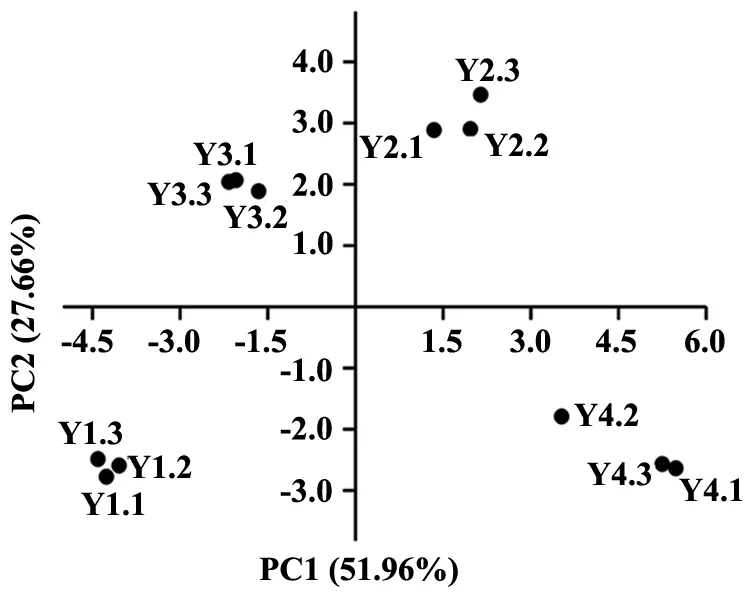
图2 土壤微生物群落的PCAFig.2 Principle component analysis of soil microbes

图3 细菌OTUs稀释曲线Fig.3 Rarefaction curves of the bacterial assemblages (OTU abundance)

样点 SampleShannon指数 Shannon indexSimpson指数 Simpson indexChao1指数Chao1 indexPD_whole_treeY110.14±0.040.0028±0.00016030.03±334.60a293.00±8.46aY29.91±0.100.0033±0.00064769.31±383.99b248.83±2.83bY310.00±0.050.0032±0.00035855.29±134.59a268.15±6.79cY410.03±0.110.0054±0.00106183.42±313.05a301.12±1.22a
同列数据(平均值±标准误)后不同小写字母者表示在5%水平上差异显著。
The data (means±SD) in the same column with the different letters mean significant differences at 5% level.
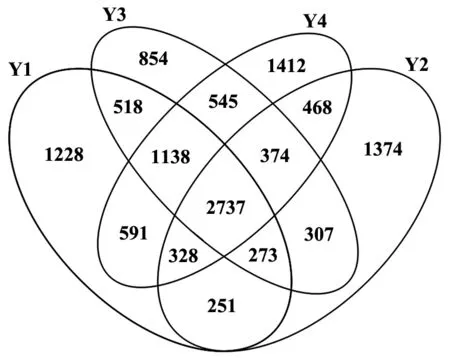
图4 细菌OTUs分布韦恩图Fig.4 Venn graph of bacteria OTUs distribution
2.4 细菌群落组成及丰度
PCA显示(图5),同一入侵区域的微生物群落在PCA图中较接近,表明同一入侵区域的微生物群落组成较相似。Y3位于PC2的正半轴,Y4位于PC2的负半轴,说明不同海拔地区的细菌物种组成有明显差异;Y1、Y2、Y3、Y4分别位于4个象限且距离较远,说明4个样点的细菌组成存在差异。
根据各OTU代表序列的物种注释结果,4种土壤样品中分别检测出细菌44门295科458属、39门269科438属、40门289科454属以及45门303科479属,其中Y2样品中检测到的细菌门数量、科数量以及属数量均低于其他3个样点,Y1和Y4土壤样品检测到的门数量不存在差异,Y1、Y3和Y4样品中所检测到的科数量和属数量不存在差异。
选取各样点土壤样品中细菌在门分类水平上最大丰度排名前10的物种,生成物种相对丰度堆积图(图6)。由图6可知,4个样点土壤样品都具有丰富的物种,在门的水平归类的细菌达到了80%以上。最大丰度排名前10的种类为:变形菌门、放线菌门、酸杆菌门、拟杆菌门、疣微菌门、厚壁菌门、浮霉菌门、芽单胞菌门、绿弯菌门以及Latescibacteria。在各采样点的土壤样品中,不同门类细菌所占的比例不同,但变形菌门的占比均为最高。
根据所有样本在属分类水平的物种注释及丰度信息,选取丰度排名前30的属,根据其在每个样本中的丰度信息,从物种和样本2个层面进行聚类,绘制物种丰度聚类热图(图7)。在属水平上,每个样点中各类细菌相对丰度存在差异,且同种细菌在不同样点中相对丰度不同。Y1中的优势菌群为固氮菌属和芽孢杆菌属;Y2中的优势菌为乳酸杆菌属、结核分枝杆菌属以及根瘤菌属和伯克霍尔德氏菌属;Y3中的优势属为鞘氨醇单胞菌属和诺卡氏菌属;而Y4样地假单胞菌属为优势菌群。

图5 细菌群落的PCAFig.5 Principal component analysis of soil bacterial communities

图6 门水平上的细菌相对丰度柱形图Fig.6 The relative abundance of bacteria in Phylum levelY1:普洱市思茅区;Y2:玉溪市红塔区;Y3:昆明市官渡区;Y4:昆明市西山区。Y1: Simao District, Pu′er; Y2: Hongta District, Yuxi;Y3: Guandu District, Kunming; Y4: Xishan District, Kunming.

图7 属水平上的物种丰度聚类热图Fig.7 The heatmap of bacterial relative abundance at the genus level
2.5 土壤的理化性质
各样品土壤养分和酶活含量均存在显著差异(表5)。Y3样点土壤样品的速效磷、铵态氮、酸性磷酸酶含量显著高于其他区域。Y2样点土壤样品的速效钾、硝态氮、有机碳、蔗糖酶、脲酶、碱性磷酸酶以及蛋白酶含量均显著高于其他样点。除铵态氮含量外,Y1样点土壤样品的其他养分及酶活含量均显著低于Y2、Y3、Y4样点。

表5 不同样点土壤样品理化性质Table 5 Physic-chemical properties of soil samples in different areas
同列数据(平均值±标准误)后不同小写字母者表示在5%水平上差异显著。
The data (means±SD) in the same column with the different letters mean significant differences at 5% level.
2.6 紫茎泽兰不同入侵区域土壤细菌群落丰度及组成与环境因子的关系
本实验中各样点土壤理化性质差异较大,为检验各土壤环境因子是否影响土壤细菌多样性及组成变化,研究采用Pearson相关性分析及Mantel test计算了细菌群落多样性指数和组成与各环境因子之间的关系。土壤因子与土壤细菌的Shannon指数和Simpson指数无显著相关,说明土壤环境因子未对4个样品的土壤细菌多样性产生影响。而土壤速效钾、铵态氮、有机碳、蔗糖酶、脲酶和蛋白酶含量与细菌群落Chao1指数呈显著或极显著相关,说明以上土壤环境因子显著影响了土壤细菌的丰富度(表6)。海拔、土壤速效钾、硝态氮、有机碳、蔗糖酶、蛋白酶以及脲酶均与细菌群落组成存在显著相关(P<0.05),即以上环境因子均显著影响了细菌群落的组成(表7)。综上所述,土壤速效钾、有机碳、蔗糖酶、脲酶以及蛋白酶是影响不同样点紫茎泽兰根际土壤细菌群落结构的主要因素。
3 讨论
土壤中的细菌是植物生长的主要驱动力之一(张丽娜等,2016),外来植物大量定居后可以通过增加与土壤养分循环有关的细菌数量来加速土壤的养分循环,提高植物根系对土壤养分的利用率,以促进自身的生长、竞争和扩散(牛红榜等,2007a)。如互花米草SpartinaalternifloraLoisel.根际土壤中细菌分布量最大,表明其在土壤中起主要作用(郑洁等,2017);薇甘菊MikaniamicranthaKunth根际土壤中细菌含量也显著高于真菌和放线菌含量(杨琼等,2015)。本实验通过PLFAs分析法和PCA,发现各样点土壤微生物中细菌含量所占比例均为最高,且细菌含量差异是导致不同入侵区域土壤微生物群落差异的主要因素。通过16S rDNA高通量测序发现,各入侵区域中土壤细菌门类数量相似,但在各区域中不同门类细菌的含量存在显著差异。其中,变形菌门、放线菌门、厚壁菌门和酸杆菌门含量相对较高,这与牛红榜等(2007b)和朱珣之等(2015)的研究结果相似。研究表明,酸杆菌门是新近基于分子生态学研究划分的新细菌类群,广泛存在于自然界各种环境中,约占土壤细菌类群的5%~46%,可能是健康土壤的指示菌(Ellisetal.,2003);变形菌门和厚壁菌门在多种植物根际土壤中也是优势类群,如葡萄VitisviniferaL.(Vega-avilaetal.,2015)、红芸豆PhaseolusvulgarisL.(Suyaletal.,2015)、菊芋HelianthustuberosusL.(Yangetal.,2016);而放线菌门多是土壤中的正常菌群(朱珣之等,2015)。

表6 环境因子与土壤细菌群落多样性指数的Pearson相关性分析Table 6 Pearson correlation analysis of environmental factors and soil bacterial community diversity index
*代表显著性P<0.05;**代表显著性P<0.01。
*indicates significant differences at 0.05 level;**indicates significant differences at 0.01 level.
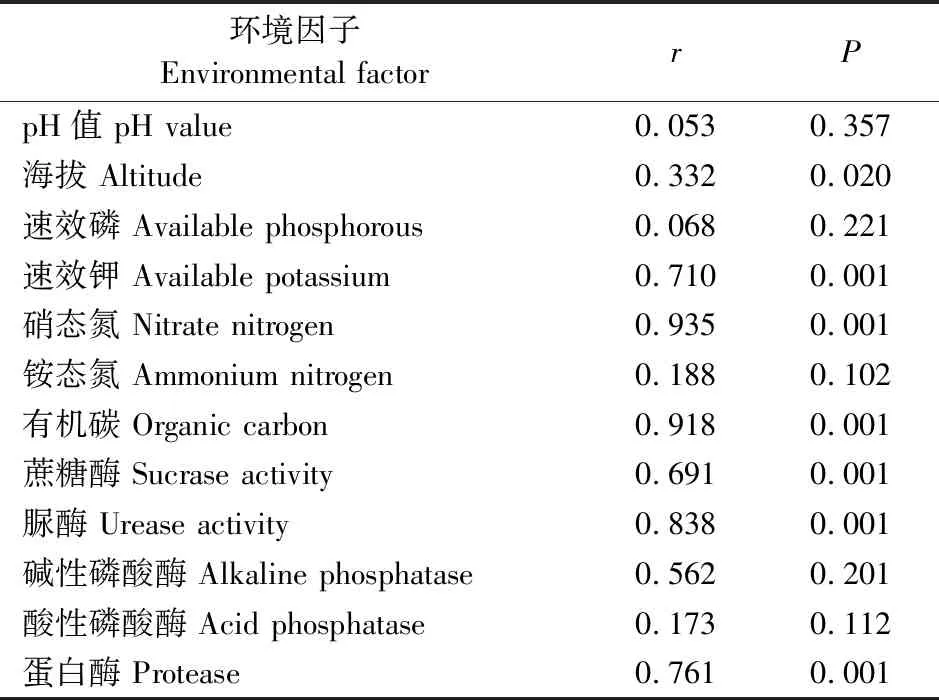
表7 细菌群落组成和环境因子的Mantel检验Table 7 Mantel tests of bacterial community and environmental factors
本实验所有土壤样品中共有的相对丰度较高的细菌为变形菌门的假单胞菌属和鞘氨醇单胞菌属,变形菌门的慢生根瘤菌属、厚壁菌门的芽孢杆菌属以及放线菌门的链霉菌属在部分区域土壤样品中相对丰度较高。牛红榜等(2007b)发现,紫茎泽兰根际土壤中存在丰富的芽孢杆菌和假单胞菌,通过这些具有强拮抗性能的根际有益微生物的反馈作用,使得紫茎泽兰能在与当地植物竞争中处于有利地位。慢性根瘤菌属、链霉菌属和鞘氨醇单胞菌属均为土壤中的有益微生物菌群(施河丽等,2018; Chaintreuiletal.,2000),在土壤碳、氮循环中起重要作用。紫茎泽兰入侵后,其根际土壤中氨氧化细菌、自生固氮菌等有益功能菌显著高于本地植物根际土壤,这些功能菌参与土壤氮循环,可间接为植物提供氮源,促进紫茎泽兰生长,使其在与本地植物竞争中获得优势(戴莲等,2012)。
土壤性质是影响微生物群落变化的主要因素之一(王光华等,2006)。本研究所选土壤人为干预较少,因此,细菌群落的变化主要体现在土壤自身理化性质的差异上。红毛草Murdannianudiflora(L.) Brenan根际土壤中的细菌含量与过氧化氢酶、蔗糖酶、纤维素酶和脲酶活性存在一定的相关性(张丽娜等,2016);而互花米草根际土壤中细菌PLFA与土壤有机碳、蔗糖酶、过氧化氢酶呈显著相关性(郑洁等,2017)。本研究4个采样地土壤样品理化性质相差较大,通过细菌多样性指数和土壤因子间的Pearson相关性分析以及细菌群落组成和环境因子的Mantel检验可知,土壤中蔗糖酶、脲酶、蛋白酶是影响紫茎泽兰根际土壤细菌群落的主要因素。土壤养分与肥力直接相关,直接影响植物生长,土壤微生物的活跃状态与土壤物质、能量循环相关,间接影响土壤肥力。杨琼等(2015)发现,薇甘菊根际土壤细菌与土壤有机质、铵态氮和硝态氮、有效磷相关性显著;邵文山等(2016)对荒漠草原4种常见植物群落土壤养分及微生物的研究发现,土壤有机质、全氮、速效磷与土壤细菌群落均存在显著正相关关系;本研究结果也显示,有机质以及硝态氮与土壤细菌群落组成关系密切。Zhangetal. (2013)指出,土壤pH值也是影响微生物多样性的主要因素,但本研究没有发现土壤pH值对土壤细菌群落及多样性有明显影响,可能与研究中涉及的样品pH值范围较近似有关(pH=6.85~7.50)。
本研究利用PLFAs分析和16s rDNA高通量测序方法相结合,比较不同采样点紫茎泽兰根际土壤细菌群落结构差异,得到如下结论:不同样点紫茎泽兰根际土壤中细菌含量存在显著差异,且细菌含量在微生物总量中占比最高。各样点土壤细菌在门类数量上相似,但不同门类的细菌相对丰度不同,其中,变形菌门的相对丰度占比均最高;从属水平上看,不同样点存在着不同的优势菌群,且同种细菌在不同样点土壤的含量差异较大。通过对土壤环境因子和细菌群落作相关性分析发现,土壤中的速效钾、有机质、蔗糖酶、脲酶以及蛋白酶是影响土壤细菌群落结构的主要因素。
