ILC细胞与自身炎症性疾病①
2019-04-12田志刚
张 彩 田志刚
(山东大学药学院免疫药物学研究所,济南 250012)
固有淋巴细胞(Innate lymphoid cells,ILCs)是一群参与固有免疫的异质性淋巴细胞,它们缺少重组激活基因(Recombination activating gene,RAG)依赖的抗原特异性受体的重排,不表达获得性免疫细胞特有的T细胞或B细胞抗原特异性受体,多位于黏膜屏障部位,接受局部微环境细胞因子的信号,通过分泌细胞因子及其他介质,发挥早期的免疫监视和免疫调节作用[1,2]。除了介导局部免疫应答外,ILCs还可作为连接天然免疫与获得性免疫的桥梁,通过协调获得性免疫细胞的功能而调节全身免疫应答[1,2]。ILCs与T细胞相互调节,放大或限制免疫应答。ILCs细胞具有可塑性,一种ILC亚群在一定条件下可能向另一群ILC亚群转化[3,4]。ILCs细胞多为组织驻留淋巴细胞,分布于肠道、皮肤、肺脏等黏膜屏障部位,参与黏膜免疫的形成、淋巴细胞的发育、组织损伤的修复及上皮屏障的保护作用[5]。一旦其数量或功能异常,将参与炎症、自身免疫性疾病、代谢性疾病、哮喘、过敏等疾病的发生发展[6,7]。本文就ILC细胞亚群的表型、发育和功能特点、与自身炎症性疾病的关系及相关治疗策略等新进展进行综述。
1 ILC细胞亚群的表型、发育和功能特点
通常根据转录因子表达、接受与分泌细胞因子的不同及发育和功能特点,将ILCs细胞分成三个亚群,即Group 1 ILCs(包括传统NK细胞和ILC1)、Group 2 ILCs(ILC2)和Group 3 ILCs(包括ILC3和LTi细胞),其中传统NK细胞具有杀伤功能,在抵抗病毒感染和抗肿瘤方面发挥重要作用,因此又称为杀伤性ILC(Cytotoxic ILCs),而其他ILCs以分泌细胞因子为主发挥功能,又称为辅助样ILCs(Help-like ILCs)[1,2]。其中ILC1、ILC2和ILC3分别对应于CD4+T细胞的Th1、Th2和Th17亚群,具有相似的细胞因子分泌谱和相似的功能,而NK细胞对应于细胞毒性T淋巴细胞(Cytotoxic lymphocytes,CTLs),具有细胞毒活性。随着对ILCs发育、转录因子调控机制的掌握及单细胞测序技术的应用,对不同ILC亚群的发育和功能特点有了更深入的解析,尤其是根据不同亚群的发育足迹,近期法国医学科学院院士、马赛吕米尼免疫学研究中心Eric Vivier教授提出,且被国际免疫学联盟批准(the International Union of Immunological Societies,IUIS)将ILC细胞分成5个亚群,即NK细胞、ILC1s、ILC2s、ILC3s和LTi 细胞[8]。
NK细胞与ILC1细胞具有许多共同的特点,如均产生以IFN-γ为主的效应细胞因子,且受转录因子T-bet的调控。但在发育路径和某些功能方面又各具不同的特点。NK细胞从共同固有淋巴祖细胞(Common innate lymphoid progenitor,CILP)经NK前体细胞(NK cell precursor,NKP)发育而来,而ILC1由CILP经固有淋巴前体细胞(ILC precursor,ILCP)发育而来[8-10]。从功能上看,NK细胞为细胞毒性自然杀伤细胞,表达高水平的穿孔素、颗粒酶等杀伤效应分子,主要功能为杀伤病毒感染细胞和肿瘤细胞,而ILC1表达低水平的穿孔素,无或仅有很弱的细胞毒活性,位于抵御病毒和胞内菌感染的第一道防线。人和小鼠的ILC1均表达CD49a和TRAIL,但活化后该标志容易丢失。ILC1常与NK细胞具有某些共同的表面标志,但又有各自不同的表型特点,如小鼠NK细胞与ILC1均表达NK1.1和NKp46等。人ILC1表达CD127,而CD16+CD56+NK细胞不表达,但外周血CD16-CD56brightNK细胞则表达CD127。CD200R曾被当作小鼠ILC1区别于NK细胞的独特标志[11]。人NK细胞表达NKp80,而ILC1不表达[12]。在转录因子表达和依赖方面,ILC1严格依赖T-bet,而NK细胞在T-bet缺失小鼠中仍然存在[13];NK细胞的发育依赖于Eomes,而ILC1在无Eomes表达时仍然正常发育。因此,Eomes表达常被认为是NK细胞的特征。
ILC2细胞高表达转录因子GATA3,在IL-25、胸腺基质淋巴细胞生成素(Thymic stromal lymphopoietin,TSLP)和IL-33刺激下主要分泌IL-4、IL-5和IL-13等Ⅱ型细胞因子。ILC2细胞具有组织驻留特性,主要分布于肠、肺、皮肤等屏障组织,既可在局部活化,在组织驻留部位发挥效应功能,又可在IL-25刺激下活化,进而通过下调CD69表达、上调S1P受体的表达,从而经血液循环到达肝、肺等远端器官[14]。这种在局部活化到远端发挥作用的特点类似于获得性免疫的特点,可以更加有效地发挥对寄生虫感染的天然免疫保护作用。炎症过后尚可通过产生双调蛋白(Amphiregulin,AREG)参与促进损伤组织的修复[15]。
ILC3细胞包括NCR+ILC3(鼠NKp46+ILC3和人NKp44+ILC3)和NCR-ILC3,其主要特点为在 IL-1和IL-23 刺激下产生Th17样细胞因子IL-17、IL-22和IFN-γ,并依赖于转录因子RORγt 和芳香烃受体(AhR)。ILC3位于黏膜部位,参与针对胞外菌和肠道共生菌的天然应答,并通过产生IL-22发挥维持肠道稳态和促进肠道干细胞增殖的作用,且调节Th17的应答。ILC3产生的IL-22可通过增强IFN-γ信号诱导肠上皮细胞产生抗菌肽(如Reg3g和Reg3b)和抗病毒蛋白,从而发挥保护作用[16]。活化的ILC3可与CD4+T细胞相互作用而调节获得性免疫应答。例如,IL-1β可刺激外周ILC3活化、分泌细胞因子、上调MHCⅡ类分子和共刺激分子的表达,发挥类似抗原递呈细胞的作用,启动CD4+T细胞的增殖和应答[17]。
LTi细胞表达LTβ、IL-2Rγ、RORγt、c-Kit和CCR6,不表达NCRs,其在胚胎发育的早期即分布至淋巴器官,对胚胎时期淋巴器官的发生及出生后淋巴结和派氏结的形成至关重要。LTi细胞通过与基质细胞相互作用,招募T、B细胞到淋巴细胞起始区域,促进淋巴器官的形成。缺失LTi细胞的小鼠(Id2或RORγt缺失)不能发育出正常的淋巴结、派氏结和肠道隐窝结节[18]。LTi细胞与ILC3相似之处为均依赖于RORγt,均能产生IL-17A和IL-22,在肠道免疫中发挥重要作用。但是,ILC3、ILC1与ILC2的分化依赖于转录因子PLZF(Promyelocytic leukemia zinc finger),而LTi和cNK细胞则不依赖PLZF。人LTi细胞表达NKp46、NKp44、NKp30等NK细胞受体。
最近,新的具有免疫调节性的ILC亚群(ILCreg或 ILC10)陆续被发现和鉴定出来,转录组学分析表明其不表达FoxP3,分泌免疫调节性细胞因子IL-10。范祖森课题组在小鼠和人的肠道中发现了一群Lin-CD45+CD127+IL-10+的ILC细胞亚群,具有明显不同于其他ILC细胞和调节性T细胞(Treg)的表型,将其命名为调节性的ILC(ILCreg)。ILCreg细胞高表达CD127、CD25、CD132、Sca-1等ILC特征性的基因以及ID2、ID3、SOX4等转录因子,但是不表达T-bet、GATA3、RORγt、FoxP3等其他类型ILC及Treg特异性的转录因子。在肠道炎症发生时,能够诱导出ILCreg的产生,ILCreg通过分泌IL-10抑制ILC1和ILC3的活化,从而抑制这两类细胞的促炎作用,在肠道炎症的转归中发挥重要的保护作用。在炎症过程中,ILCreg细胞能够分泌TGF-β,并以TGF-β自分泌的形式维持其自身的增殖,促使其更好地发挥肠道保护作用[19]。另一群以分泌IL-10为主的肺脏ILC2(命名为ILC210)具有相似的免疫调节特性,能够通过分泌IL-10抑制肺部的病理性炎症应答。ILC210表达比ILC2更高水平的Id3、Foxf1、Atf3和Klf2,不表达FoxP3[20]。
2 ILCs细胞在炎症、组织稳态和修复中的作用
ILCs主要位于非淋巴组织,尤其是黏膜屏障部位,能够及时感受炎症、危险或损伤信号,进行自我更新和针对环境刺激做出应答,在调节炎症、维持或重建组织稳态、促进损伤组织修复、维持或重建黏膜屏障(包括肠道、呼吸道、肺、皮肤等组织)等方面发挥重要作用[21]。例如,ILC能够与上皮细胞相互作用共同维持上皮的稳态。IL-33能够促进ILC2分泌上皮生长因子AREG,AREG为表皮生长因子受体(EGFR)的配体,可作用于肠道上皮细胞,促进上皮的生长和分化,维持肠道上皮的稳态,防止肠道炎性肠病(Inflammatory bowel diseases,IBD)的发生[15,22]。同样,肺脏气道上皮细胞通过分泌IL-33促进ILC2的活化,如果缺失ILC2则影响上皮细胞的损伤修复,且该修复作用依赖于AREG[23]。IL-9可促进ILC2的存活、AREG的产生和肺组织的修复[24]。ILC2亦以IL-33依赖的方式促进皮肤伤口的愈合[25]。位于肠道固有层的ILC3参与维持肠道组织的稳态,尤其是其产生的IL-22在维持黏膜组织稳态平衡和肠道黏膜屏障的完整性方面起关键作用。正常情况下,IL-22的分泌会促进上皮稳态的维持,防止共生菌结构紊乱和慢性炎症的发生。IL-22能够促进肠道隐窝处的肠道干细胞的增殖,促进异基因骨髓移植后的肠道上皮再生,并保护肠上皮细胞免受T细胞介导的杀伤,降低急性移植物抗宿主病(Graft-versus-host disease,GVHD)导致的肠道病理损伤和死亡率[26]。肠道组织损伤刺激NKp46+和CCR6+ILC3s的活化,活化的ILC3可通过分泌IL-22促进肠道干细胞STAT3磷酸化,抑制肠道干细胞的凋亡;同时,促进肠道干细胞的增殖,进而缓解放、化疗对肠道干细胞的损伤[27]。
3 ILCs与自身炎症性疾病
ILCs对组织的过度修复、长期的慢性刺激使ILCs的保护性应答转变成病理性应答,进而导致严重的炎症性疾病和组织的纤维化增生。在自身炎症性疾病的发生发展过程中,可以理解的是ILC1和ILC3通常参与炎性肠病或感染诱导的肠炎等疾病的病理过程,而ILC2通常参与过敏性疾病的发病过程。但在不同的疾病模型、不同的发病阶段及不同的组织微环境下,ILC亚群可能发挥双向作用,而且ILCs的可塑性使不同的ILC亚群之间可发生转换,从而发挥不同的效应功能(图1)。
3.1ILCs与肠道炎症相关性疾病 肠道黏膜和相关淋巴组织被认为是人和小鼠ILCs细胞的储存库。在稳态情况下,ILCs占小肠固有层淋巴细胞的10%。各ILC亚群在肠道均有分布,但ILC3占比例最多,且在维持肠道稳态方面发挥重要作用[28]。正常情况下,ILC3产生IL-22、ILC2产生AREG促进维持肠道上皮屏障的完整性、对共生菌的限制和损伤后的组织再生。清除ILC3或缺失IL-22会导致共生菌产碱杆菌的播散,使之定殖至淋巴器官,进而启动慢性炎症[29];IL-22缺失会加重DSS诱导的肠炎。肠道环境中的前列腺素E2(PGE2)与ILC3表面的受体EP4结合,触发IL-22的分泌。然而,研究发现,ILC3通过产生IL-17、IL-22和IFN-γ亦是肠道炎症的主要介导者。的确,IBD患者肠道ILC3的数量和活性明显高于健康对照者;而ILC3缺失小鼠或通过抗体清除ILCs细胞可明显降低肠炎的病理评分。同样,IBD小鼠和患者肠道ILC1细胞的数量和活性亦明显升高[30]。人肠道ILC1分为固有层ILC1(表达CD161和CD127,但不表达NKp44)和上皮内ILC1(表达NKp44和CD103,但不表达CD127)两个亚群,ILC1细胞的数量(尤其是固有层ILC1)在克罗恩病患者肠黏膜明显增多,且与疾病的严重程度成正相关[6]。在炎症情况下,IL-12可使ILC3下调RORγt表达,获得ILC1样的特性(称为ex-ILC3 ILC1),并与ILC1一起通过产生IFN-γ、IL-17A和GM-CSF介导组织的免疫损伤,并导致巨噬细胞的活化和中性粒细胞的浸润。值得注意的是,ILC3与ILC1之间的转化是可逆的,ex-ILC3 ILC1细胞在IL-23、IL-1β和维甲酸的作用下又可恢复到ILC3的表型。ILC3与ILC1的相互转化由肠道固有层的专职DC控制,其通过提供细胞因子微环境调控ILCs的命运[28]。有趣的是,有研究发现ILC2亦可转化为分泌IFN-γ的ILC1,在克罗恩病患者的肠道组织中数量增多,而ILC1在IL-4的作用下亦可转化为ILC2。总之,肠道ILCs细胞,尤其是ILC3,在肠道稳态和炎症情况下发挥保护和促炎的双向作用,这种ILC亚群之间相互转化的机制可被用于炎症性肠病患者治疗的靶点和策略。
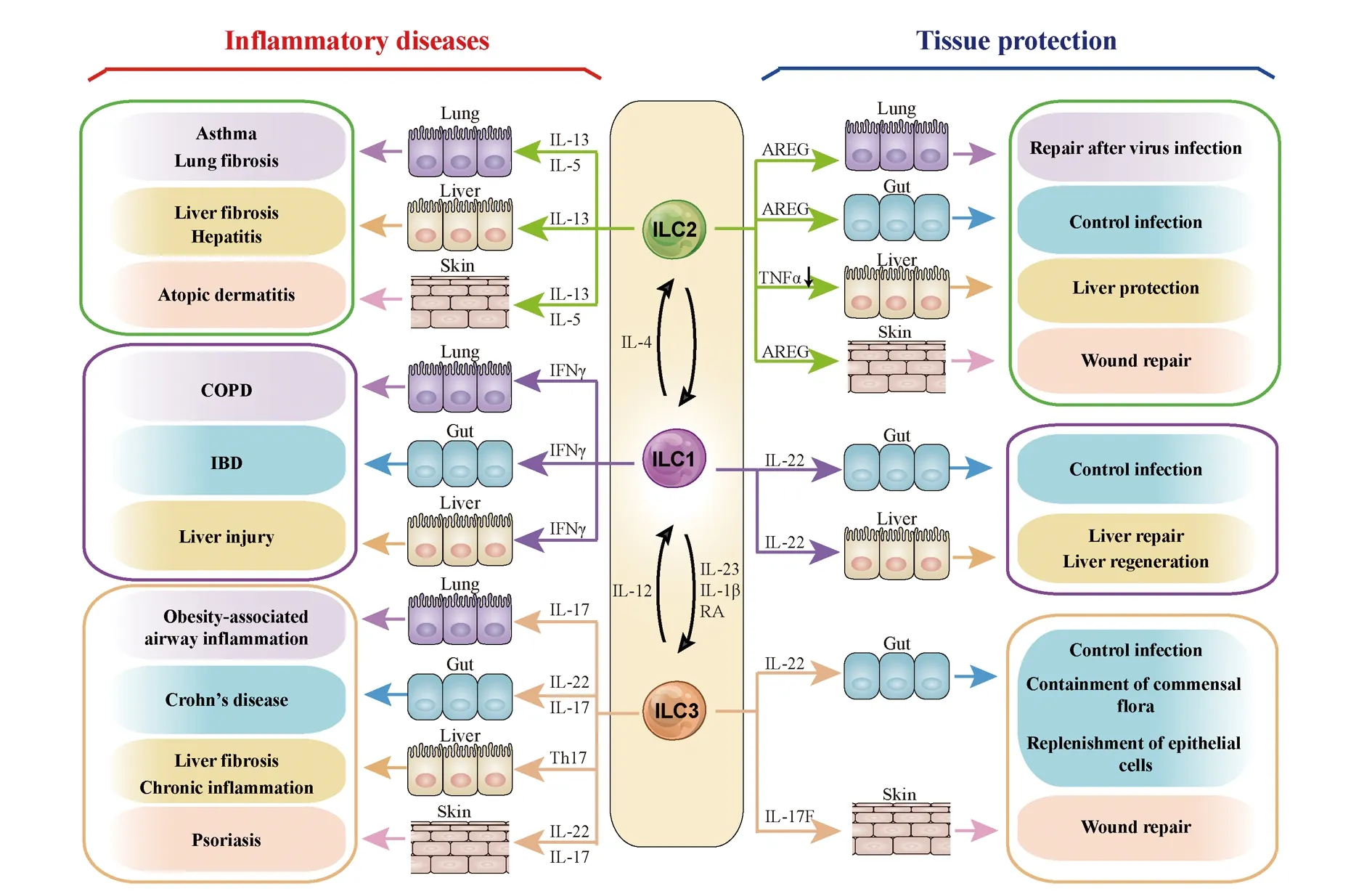
图1 ILCs细胞发挥组织保护和促进炎性疾病发生的双向作用Fig.1 Innate lymphoid cells: dual roles in inflammatory diseases and tissue protection
3.2ILCs与肺脏炎症相关性疾病 NK细胞是人和小鼠肺ILCs细胞的主要亚群,占人肺总ILCs细胞的95%。除NK细胞外,人ILC3(主要为NKp44-ILC3)为肺部主要的ILC亚群,而小鼠ILC2在稳态时占的比例相对较多[31]。NK细胞在肺脏黏膜组织发挥重要的免疫防御、免疫监视和免疫清除作用。肺组织分布的ILCs细胞通过感受危险信号,活化、增殖、分泌细胞因子,参与肺黏膜组织区域的免疫监视、免疫调节、损伤后的组织修复和稳态的维持。但是,一旦失控,就会导致病理损伤、炎症和疾病的发生。
ILC2是参与促进肺部过敏反应、炎症和纤维化形成的主要ILC细胞亚群[6,31]。哮喘患者的外周血和肺泡灌洗液中ILC2细胞的数量明显增多,且肺泡灌洗液中ILC2细胞的数量和IL-33水平与哮喘患者呼吸功能衰竭的程度相关。肺上皮来源的IL-25、IL-33和嗜碱性粒细胞产生的IL-4刺激ILC2的活化,促进肺部和皮肤的炎症,并参与过敏性疾病(如哮喘、慢性鼻窦炎)的发生。呼吸道合胞病毒或鼻病毒感染会诱导分泌IL-13的ILC2的活化与增殖,进而破坏支气管上皮的完整性[32]。ILC2还与其他细胞相互作用,促进气道炎症的发生与进展[6]。例如,嗜碱性粒细胞来源的IL-4及ILC2细胞间通过ICOL-ICOL相互作用均可促进ILC2的活化。IL-33激活的肥大细胞可扩增Treg细胞进而抑制ILC2的活化;然而在IgE交联和脱颗粒后,肥大细胞又可通过释放脂质介质[如白三烯D4和前列腺素D2(PGD2)]驱动ILC2的活化。白三烯D4和PGD2还可促进ILC2到肺脏气道炎症部位的聚集,并促进Th2细胞细胞因子的产生[33,34]。在过敏性哮喘模型中,ILC2与Th2细胞通过CD40L和MHCⅡ类分子相互作用,促进Th2的活化;表达MHCⅡ类分子ILC2还可与抗原特异性CD4+T细胞相互作用,T细胞产生的IL-2促进ILC2的增殖和IL-13的产生[35]。ILC2分泌的IL-13促进活化的肺脏树突状细胞(Dendritic cell,DC)迁移至引流淋巴结,激发naïve T细胞分化成Th2细胞;还可促进IRF4+CD11b+CD103-DC产生招募Th2的趋化因子CCL17[36,37]。ILC2加重肺脏炎症会进一步促进肺纤维化的发生。IL-33诱导ILC2和Ⅱ型巨噬细胞(M2)的扩增,ILC2进一步分泌IL-13促进M2的极化和纤维母细胞的活化,加重纤维化的进程[38]。
在肥胖诱导的哮喘中,ILC3通过产生IL-17参与调节由炎症小体活化所产生的IL-1β介导的气道超敏反应[39]。ILC1在慢性阻塞性肺病患者肺和外周血中比例增多,且与疾病严重程度相关。吸烟、细菌或病毒感染促进肺部ILC2向ILC1转变,促进慢性阻塞性肺病的发生发展[40]。
3.3ILCs与肝脏炎症相关性疾病 肝脏富含NK细胞,NK细胞占小鼠肝脏淋巴细胞的30%,其中50%为传统的NK细胞(cNK),另50%为肝脏驻留NK(LrNK)细胞。LrNK细胞具有与黏膜ILC1相似的发育途径,被认为是肝脏ILC1细胞,表型为CD49a+CD49b-,高表达TRAIL和FasL[41-43]。肝脏NK细胞和ILC1细胞在肝脏炎症相关性疾病中发挥保护或促炎双向作用[44]。一方面,NK通过杀伤活性和分泌细胞因子、趋化因子的能力在肝脏免疫防御、清除病毒感染和抗肿瘤方面发挥重要的作用;而且,肝脏cNK细胞和ILC1均可通过分泌IL-22修复肝脏损伤、促进肝脏再生。另一方面,肝脏NK和ILC1细胞亦通过促进肝损伤而发挥有害的作用,例如,肝脏cNK细胞因为增强肝脏CD8+T细胞的活性而在HBV感染和Con A诱导的肝损伤过程中发挥促进肝损伤的作用。慢性HCV感染中肝脏ILC1高表达NKG2A,抑制cNK和CD8+T细胞的抗病毒活性,而发挥促进肝脏免疫耐受的作用,阻断NKG2A信号则增强cNK和CD8+T细胞中IFN-γ的分泌,促进HCV的清除[45]。
肝脏应激或炎症状态下诱导IL-33产生,进而导致肝脏ILC2的活化和聚集,活化的ILC2产生IL-13促进肝星形细胞的活化,从而加重炎症损伤和促进肝纤维化的形成[46]。在Con A介导的肝炎模型中,CD4+T细胞活化促进IL-33的释放,进而活化并促进肝脏ILC2分泌IL-13和IL-5,后者进一步募集嗜酸性粒细胞到肝脏聚集,放大炎症反应[47]。但是,亦有基于IL-33缺失或其受体ST2缺失小鼠的研究认为肝脏ILC2在肝损伤中起保护作用[48]。在腺病毒介导的肝炎模型中发现IL-33扩增活化的ILC2可通过抑制TNF-α的产生而发挥肝脏保护作用[49]。
多项研究认为ILC3在CCl4、Con A和酒精诱导的肝损伤中通过抑制IFN-γ表达、诱导IL-22分泌而发挥保护作用[44,50],但是在HBV感染研究中发现IL-22可通过促进Th17细胞聚集到肝脏而加重肝脏的慢性炎症和纤维化进展[51]。不同的结论可能与观察的模型、疾病进展的阶段不同有关,确切的机制尚有待更深入的探讨。
3.4ILCs与皮肤炎症相关性疾病 稳态时不同ILC亚群在人和小鼠皮肤均存在。ILCs在皮肤损伤修复和稳态维持方面发挥重要作用[6,52]。在皮肤损伤时,ILC3对表皮Notch1应激信号做出应答,向伤口部位聚集,并分泌IL-17F。ILC3缺失会导致表皮细胞增殖和伤口愈合的延迟。IL-33-ILC2轴在损伤后皮肤屏障的恢复方面亦起重要作用[53]。
ILC2参与皮肤的炎症病理过程。已发现在过敏性皮炎等多种皮肤炎性疾病的皮损部位ILC2的数量和比例明显高于正常皮肤组织,ILC2的激活剂TSLP、IL-33和IL-25表达上调,且ILC2表达IL-25受体、IL-33受体ST2、TSLP受体和PGD2的受体CRTH2的水平明显升高[54]。清除ILC2可明显减轻皮肤损伤和炎症程度。ILC2还可与皮肤部位其他细胞相互作用调节皮炎的发生发展。例如,ILC2与嗜碱性粒细胞(分泌IL-4)或肥大细胞(分泌PGD2)相互作用,可促进炎症的进展;而活化ILC2表达的KLRG1与皮肤角质细胞的E-cadherin相互作用可抑制ILC2的活性,对炎症应答起负反馈的调节作用[54]。另一项研究则发现,过敏性皮炎部位的角质细胞高表达NKp30的活化配体B7-H6,与ILC2表面的NKp30结合可促进ILC2分泌IL-5和IL-13,用抗体阻断NKp30或给予抑制性配体galectin-3则阻断Ⅱ型细胞因子的分泌[55]。
ILC3参与牛皮癣的皮肤炎症发病过程。牛皮癣患者的炎性皮肤部位含有大量的ILC3细胞,且能够分泌IL-22和IL-17A。在咪喹莫特(Imiquimod)诱导的牛皮癣模型(能够模拟人牛皮癣斑块形成)中,浸润至皮肤部位的T细胞和RORγt+ILC3细胞(而非Th17细胞)为IL-17A、IL-17F和IL-22的来源,这些细胞和因子是牛皮癣皮肤炎症发生必需的、足以促其发病的原因[56]。
4 展望
经过十余年的研究,目前科学家们已对ILCs细胞的分类、发育分化、细胞和生物学功能及与不同疾病的关系有了深入的理解。调节性ILC亚群的发现进一步完善了ILCs家族成员,但尚需进一步探讨除肠道外其他黏膜屏障组织(如肺、肝、皮肤等)是否存在ILCreg以及是否在自身炎性疾病中发挥调节作用。记忆性NK细胞和记忆性IL-7R+ILC1的发现进一步验证了天然免疫细胞亦具有长期记忆性应答的特性[57,58]。
鉴于不同ILC亚群在免疫监视、免疫防护、组织修复、稳态维持和炎症应答中的重要作用,ILCs细胞有望成为免疫治疗或炎症相关疾病治疗的靶点。通过激活ILCs活性可增强机体对病原体感染的抵抗和清除作用或促进组织的修复,而抑制或阻断ILCs的活性或其产生的效应分子有助于慢性炎症性疾病的治疗。例如,IL-2、IL-12和IL-18可激活ILC1,IL-25和IL-33可激活ILC2,IL-1β和IL-23可激活ILC3;而相应细胞因子的抗体则可抑制ILCs的活性,用于相应炎性疾病的治疗。RORγt抑制剂可限制ILC3的促炎症作用,抑制RORα的活性可能影响ILC2的功能。最近的一项研究用小鼠骨髓移植模型证明过继转输ILC2可防治GVHD,使除NK细胞外,以ILC亚群进行细胞治疗成为可能[59]。记忆性NK和ILCs的发现为开发基于ILCs的疫苗带来了希望[57,58]。但是,由于ILCs与T细胞在表达的转录因子、分泌的细胞因子、发挥的功能等方面存在很多相似之处、ILCs细胞自身功能的双向性、ILCs的异质性和不同亚群之间的相互调节和转化等,均导致目前能够采取的治疗策略缺乏特异性。迫切需要找到ILCs及其亚群特异性的标志,或找到针对ILCs及其不同亚群的特异的活化剂或抑制剂,才可能以这些特异性的标志为靶点或特异性清除或转输某一特定ILC亚群,而达到治疗相应疾病的目的。
