FGA基因c.104G>A杂合突变导致的先天性低纤维蛋白原血症家系的基因分析
2019-04-12苏正仙毕晓洁金先富张慧斐姜俊宇沈波
苏正仙 毕晓洁 金先富 张慧斐 姜俊宇 沈波
纤维蛋白原(FIB)是参与机体止血的关键物质,具有多种功能,包括纤维蛋白凝块的形成、与凝血酶的结合、介导血小板聚集反应等[1]。它存在于血浆中,由肝细胞分泌,分子质量为340kD,是通过29个链间和链内二硫键连接的两组三个多肽(Aα、Bβ和γ)组成的完全六聚体[2]。六条链排列形成三叉结构,通过由包含两个单独三螺旋卷曲螺旋区域的2个远端球形D结构域以及一个中心E区域组成。每条链的N末端有助于构成中心E区域,而Bβ和γ链的C末端和卷曲螺旋区域的一部分形成2个远端球形D结构域[3]。形成FIB的3条多肽链(Aα、Bβ 和 γ)分别含有 610、461以及 411个氨基酸残基,每条多肽则分别由相对应的不同基因参与编码翻译产生[4]。FIB基因缺陷可影响其FIB的表达,或明显影响其表达产物与血小板糖蛋白、血浆凝血酶之间的结合位点,更有可能影响到FIB单体的聚合及其蛋白的稳定性,由此最终导致机体表现出与出血、凝血相关的一系列少见的病症[5-6]。笔者回顾1例遗传性低纤维蛋白原血症患者家系的临床资料,并通过基因测序来对其家系的发病机制进行初步研究。
1 资料和方法
1.1 一般资料 本例患者来自浙江台州地区的先天性低纤维蛋白原血症家系。先证者,男,49岁,患痔疮、反复渗血前来本院就医,发病原因不明。2016年5月患者行凝血功能检查,结果FIB为0.68g/L,血常规显示为缺铁性低色素性贫血,且在1个月后凝血功能复查时FIB值仍处于严重低值,结合D-二聚体与FIB降解产物(FDP)结果,排除了原发及继发性纤溶亢进后,结合家系咨询,初步诊断为先天性低纤维蛋白原血症。未发现该患者有肾脏疾病、肝炎肝硬化的发病历史;此外其家族内部也从未有患肿瘤的病史等。为求进一步确诊,笔者对该家系进行了基因测序。其家系图谱见图1。
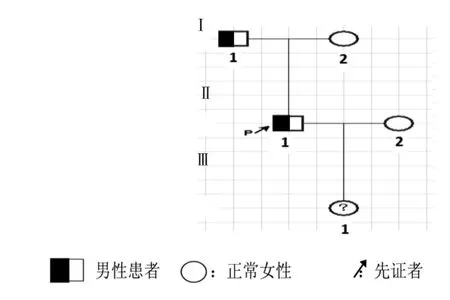
图1 先证者家系图谱
1.2 方法
1.2.1 引物 根据FIB的基因序列设计29对引物[7],包含FGA、FGB以及FGG基因的所有外显子及其侧翼序列和启动子区,并送上海华大基因公司进行合成。
1.2.2 标本收集 采血前与其家系成员签署相关的知情同意书。采集家系各成员外周静脉血样本两份,一份按照1∶9的比例以0.109mmol/L的枸橼酸钠抗凝,行凝血相关数据的检测;另一份常规血清分离,用于DNA的抽提,血样本放置于-80℃的冰箱中冰冻保存。
1.2.3 凝血功能检测与基因测序 将枸橼酸钠抗凝的血样以3 000r/min离心10min,上层血浆运于检测APTT、PT、FIB、TT、D-二聚体、FDP(法国 STA-R 全自动血凝分析仪以及其配套试剂),FIB抗原含量采用免疫比浊法测定(雅培c16000全自动生化分析仪及其配套试剂)。另将分离出的血清用于肝肾功能检测(雅培c16000全自动生化分析仪以及其配套试剂)。用采集的全血标本进行DNA抽提,经PCR扩增后外送上海华大基因检测公司进行测序,测序结果与NCBI GenBank中FIB所对应的基因测序数据做最终地比较分析,并用反向测序予以验证突变位点。
1.2.4 生物信息学工具 采用Polyphen-2(蛋白质ID:P02671)、MutationTaster(蛋白质转录本 ID:ENST00000 302053)两种在线软件分析FGA基因第2外显子c.104G>A的错义突变对FGA蛋白结构和功能的影响。
2 结果
2.1 肝功能、凝血项目检测结果 先证者家系成员的肝功能项目检查结果均显示处于正常范围,排除肝脏功能合成障碍引起的FIB缺陷。见表1。
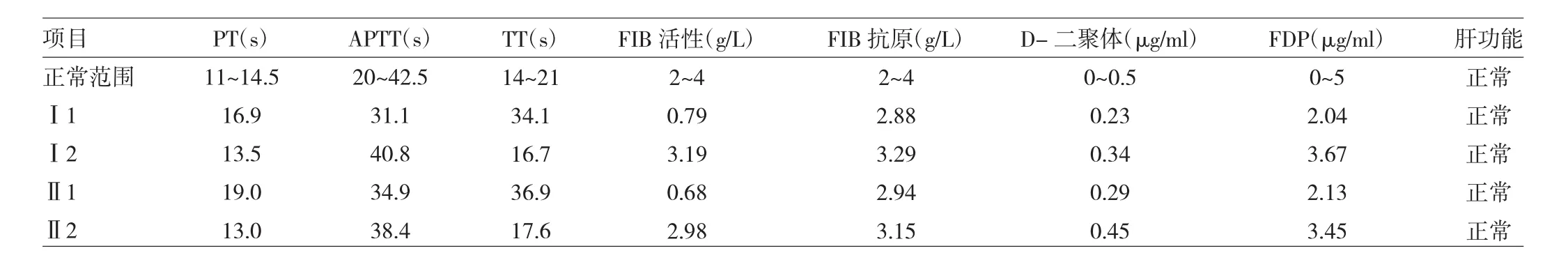
表1 家系成员的检测结果
2.2 基因检测结果 基因测序结果见图2,先证者FIBFGA基因第2外显子发生c.104G>A的碱基杂合点突变(密码子CGT→CAT),导致Arg16His的杂合突变(16号精氨酸突变为组氨酸)。先证者的父亲在该位点的测序结果与先证者一致,除此以外所有其它的基因测序结果都显示为正常。
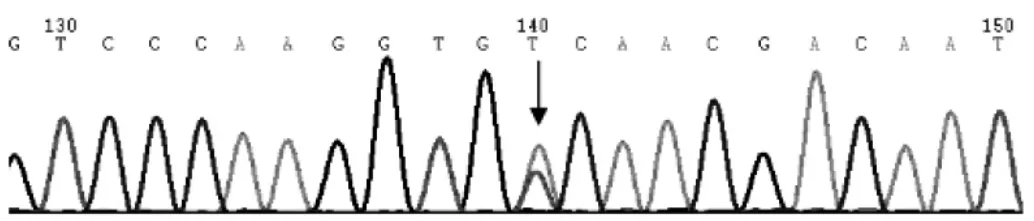
图2 先证者FGA基因第2外显子测序图
2.3 生物信息学结果 两种在线软件预测结果一致,Pyolphen积分为1,预测结果为“PROBABLY DAMAGING”,MutationTaster积分为 0.9999,预测为“disease causing”。
3 讨论
FIB是机体中含量最高的凝血因子,其在血浆中的正常浓度范围为2.0~4.0g/L[8]。当外界创伤等引起血管出血时,FIB就会在血浆一系列凝血因子及相关酶类的催化作用中转变成为纤维蛋白,大量纤维蛋白纵横交叉呈网状,结合血小板形成微血栓达到止血的目的[9],不同原因导致的FIB含量或者活性降低都有可能会导致机体出凝血功能上的一系列障碍,引起出血难止亦或者纤溶激活障碍导致血栓栓塞,最终可能危及生命。
20世纪终末期,Terasawa等[10]首次发现低纤维蛋白原血症存在的具体基因缺陷-杂合的γCysl53Arg,之后陆续有文章报道新发现的突变位点,截止2017年1月人类基因突变数据库已发现近300个突变位点,而FGA基因上就存在100多个突变位点,包括错义/无义突变、剪切位点突变、小片段缺失等。
遗传性低纤维蛋白原血症临床表现具有较大的异质性[11],严重程度与FIB的突变位点及类型相关,同一突变也有可能有不同临床表现。Stucki等[12]研究显示Arg16His突变者无出血症状。而本文研究家系,先证者表现为痔疮、反复渗血,FIB为0.68g/L,明显低于正常水平,基因测序结果显示FGA基因2号外显子上存在c.104G>A的错义突变(密码子CGT→CAT),即Arg16His的杂合突变。这一突变不仅可以导致FIB被凝血酶酶解时FPA释放速度的减慢[13],更严重的可能是此突变还可引起FIB被酶解过程中产生FPA总量的异常,凝血过程中交联纤维蛋白产生量因此减少,导致FIB在机体血浆中的“量”和活性先天性下降的现象,本文推测导致该突变是导致该家系先证者反复渗血的主要原因。
综上所述,本研究明确了FGAArg16His的突变是导致本研究家系遗传性低纤维蛋白原血症发生的分子机制。但是这其中所蕴含的具体机制则还需要进行深入的研究探索,对造成先天性低纤维蛋白原血症的分子机制也同样需要进一步地被阐明。目前,国内在先天性低纤维蛋白原血症的相关研究取得了很大的进展,随着检测技术的逐渐进步完善,可以通过分析纤维蛋白溶解曲线、FIB凝固率检测等方法[14]较准确地评估FIB功能,以此分析突变与疾病之间的关系[15]。目前该类疾病的治疗仍以替代治疗为主,基因治疗是未来的发展趋势[16]。
