采用浸酸牛皮提取胶原及其性质表征
2019-04-10田振华
田振华, 李 幸, 王 浩
(1.陕西科技大学 轻工科学与工程学院 轻化工程国家级实验教学示范中心, 陕西 西安 710021; 2.中国皮革和制鞋研究院, 福建 晋江 362201)
0 引言
制革是指将生皮鞣制成革的过程.在制革过程中会对牛皮进行浸灰、浸酸等鞣前处理,去除毛和非胶原成分并适度松散胶原纤维,以便于促进鞣剂、加脂剂等化料的均匀渗透.通常,胶原在强酸(硫酸等)、强碱(石灰、氢氧化钠等)长时间作用时,胶原分子内的氢键会发生断裂、肽键可能会被水解,从而导致胶原二级结构(三股螺旋结构)的崩塌.
对于强酸、强碱对胶原二级结构的影响,已有研究表明浸灰过程所使用的强碱对裸皮中胶原分子三股螺旋结构没有太大影响[1];对于浸酸过程,Chen等[2]在不浸酸铬鞣剂C-2000的应用研究中提到在牛皮的浸酸过程中,皮胶原和许多酸溶性、盐溶性蛋白质会有所损失,但未进一步研究胶原分子三股螺旋结构的变化.因此,探究浸酸过程对胶原的结构与物化性质方面的影响具有重要的意义.
更为重要的是,制革原材料——生皮的主要成分胶原也是一种非常热门的天然高分子材料,因具有良好的生物相容性、低抗原性、可生物降解性,因此广泛应用于医学、化妆品、生物材料、功能食品等领域[3].目前,针对牛皮胶原的提取主要采用的原材料有鲜皮与灰碱皮[1,4].其中,鲜牛皮需经刮毛、去肉、脱毛、脱脂、脱色等预处理去除非胶原成分,操作工序复杂、提取时间较长;灰碱皮提取胶原需要先进行脱灰脱钙处理,对钙残留量要求较高,且灰碱皮本身无法长期储存[5];而对于浸酸皮,只需中和、水洗后便可用于胶原的提取,前处理工艺简单、需时较短且浸酸皮便于保存.相比之下,从浸酸牛皮中提取胶原是一种相对便捷的方案.有专家针对酸肿皮即浸酸牛皮遇水充水膨胀、胶原纤维水解的裸皮进行了研究,结果发现胶原分子的成纤维能力明显下降且所得纤维数量少、不成熟,所以酸肿皮作为鲜皮的替代物用于提取胶原可行性有限[6].
综上,本文拟采用浸酸牛皮作为原材料提取胶原,并与鲜牛皮胶原进行对比,探究浸酸过程对胶原二级结构的影响,并评估浸酸牛皮是否可作为原材料用于胶原提取的可行性.
1 实验部分
1.1 试剂与仪器
(1)主要试剂:冰乙酸、氯化钠、碳酸钠等,分析纯,广东光华科技有限公司;胃蛋白酶,生化级,如香生物科技有限公司.
(2)主要仪器:傅里叶变换红外光谱仪,德国Bruker,Vector-22;微量差示扫描量热仪,英国马尔文,MicroCal VP-Capillary;旋转粘度计,美国Brookfield,DV-II;电泳仪,美国Bio-rad,Protean Tetra;原子力显微镜,美国Agilent,AFM 5100.
1.2 鲜牛皮胶原和浸酸牛皮胶原的提取
1.2.1 牛皮的预处理
新鲜牛皮经刮毛去肉、水洗后,采用灰碱处理24 h脱毛并去除部分非胶原成分,进一步去肉后将皮块剪碎,水洗脱灰直至pH约为5 ~ 6.随后采用脱脂剂进行脱脂24 h.随后,采用7.5% NaCl溶液浸泡以除去盐溶性非胶原成分,水洗除盐后备用.
对于浸酸牛皮,只需采用Na2CO3中和,调节pH约为5 ~ 6,剪碎皮块并水洗除盐后备用.
1.2.2 胶原的提取与纯化
将预处理好的两种碎皮分别置于含有3%胃蛋白酶(EC 1∶3000)的0.5 mol/L醋酸溶液中,搅拌48 h后低温高速离心10 min,取上清液.经多次盐析、离心、溶解、透析后得到两种纯化胶原溶液,最后将胶原溶液冷冻干燥,冻干胶原海绵于干燥器中保存备用.鲜皮胶原和浸酸皮胶原分别命名为FS和PS.
1.3 胶原的理化性质检测
1.3.1 十二烷基磺酸钠-聚丙烯酰胺(SDS-PAGE)凝胶电泳
按照Laemmli[7]的SDS-PAGE方法分析胶原的分子量分布情况.将一定量的胶原与样品处理液(Tris-HCl缓冲液、甘油、SDS等)混合均匀,煮沸5 min后冷却备用.选用7.5%的分离胶和4%的积层胶进行电泳,分离结束后染色、脱色直到蛋白质条带清晰为止.
1.3.2 紫外分光光谱分析
将FS和PS分别溶于0.1 mol/L醋酸溶液中,制备浓度为1 mg/mL的胶原溶液.以0.1 mol/L醋酸溶液作空白,在190~600 nm波长范围内进行紫外扫描.
1.3.3 傅里叶红外分析
将FS和PS溶液分别于室温下倒入聚四氟乙烯盘、自然干燥成膜.为避免胶原膜中醋酸的影响,使用超纯水对胶原膜进行浸泡,清洗除去醋酸,自然干燥后置于干燥器中平衡水分.采用傅里叶红外光谱仪获取红外谱图.测定范围为400~4 000 cm-1,分辨率4 cm-1,扫描信号累计64次.
1.3.4 表观粘度
将胶原海绵溶解于0.1 mol/L醋酸溶液中,制备浓度为2 mg/mL的胶原溶液.在室温下,采用旋转粘度计监测胶原溶液的表观粘度与剪切速率的关系曲线.测试温度为25±0.1 ℃,剪切速率的范围为0.01~1 s-1.采用以下三种模型对所得到的表观粘度数据进行拟合.
(1)Ostwald-de Waele模型:
(1)

(2)Carreau模型[8]:
(2)
式(2)中:η0和η∞分别指零剪切粘度和无穷剪切粘度(Pa·s);λ是特征时间;m是材料常数,相当Ostwald-de Waele模型中的参数n,无量纲.
(3)Cross模型[9]:
(3)
式(3)中:k和a是材料常数.其中,a相当Ostwald-de Waele模型中的参数n,无量纲.
1.3.5 微量差示扫描量热分析(US-DSC)
将FS和PS分别溶于0.1 mol/L醋酸溶液中,制备浓度为0.5 mg/mL的胶原溶液.以0.1 mol/L醋酸溶液作空白;升温速率为1 ℃/min,温度范围为25 ℃~60 ℃,绘制吸热曲线并对吸热峰进行积分计算得到样品的焓变值(ΔH).
1.3.6 成纤维动力学测试
将冻干胶原海绵剪碎后溶于磷酸盐缓冲液(10 mmol/L Na2HPO4/NaH2PO4+100 mmol/L NaCl,pH7.4)制备1 mg/mL的胶原溶液.将胶原溶液置于37 ℃恒温水浴中,记录60 min内其在313 nm处的吸光度数值.
1.3.7 原子力显微镜(AFM)观察
将15μL 1.3.6中所得到的胶原纤维滴加在新剥离的云母片上,使其自然铺展,自然干燥24 h.使用AFM观察样品的表面形貌.采用软件ImageJ 1.44测量胶原纤维的直径并绘制直径分布图.
2 结果与讨论
2.1 胶原的分子量分布分析
图1为PS与FS的外观图及电泳图.由图1(a)可知,两种胶原溶液均呈现澄清透明.由SDS-PAGE谱图1(b)可知,两种胶原的电泳条带类似,均有三条不同的条带.对照蛋白质标准样可知,分子量约在100 kDa附近的两条电泳条带分别对应胶原的α1链和α2链,在200 kDa附近的条带为胶原的β链.由条带的深浅程度可推测α1链浓度高于α2,并且PS与FS电泳谱图类似,保留了胶原的三股螺旋结构.此外,电泳图中除两条α带和一条β带外无其余小分子条带,说明所提取的胶原中无小分子水解胶原或杂蛋白[10].综上可知,所提取的胶原主要为I型胶原且纯度较高.

(a)外观图 (b)电泳图 图1 FS和PS的外观图与SDS-PAGE 电泳谱图
2.2 紫外图谱分析
图2是FS和PS的紫外吸收光谱图.蛋白质的紫外光谱图实际上是蛋白质分子的各种紫外生色基团加和的结果,大多数蛋白质在280 nm处有最大紫外吸收,然而胶原不含有色氨酸,故在280 nm处无明显紫外吸收峰[11].FS和PS的紫外吸收峰分别在223 nm和218 nm处,均在胶原的特征紫外吸收峰215~230 nm范围内,表明从浸酸牛皮、鲜牛皮中提取的产物是胶原,该吸收峰的出现主要是由于肽链中C=O键的n→π*跃迁产生的.
2.3 红外图谱分析
图3是FS和PS的红外(FTIR)图谱.胶原的FTIR图谱主要来自于酰胺基团的贡献.根据标准手册与相关文献[12],对PS和FS的特征吸收峰进行指认.其特征吸收峰主要包括在3 320 cm-1附近归属于N-H伸缩振动和氢键耦合的酰胺A带、来源于C-H伸缩振动在3 080 cm-1处附近出现的酰胺B带、1 658 cm-1和1 653 cm-1处的酰胺Ⅰ带(C=O伸缩振动和C-N伸缩振动)、在1 552 cm-1和1 554 cm-1处归属于N-H伸缩振动和C-N伸缩振动的酰胺Ⅱ带、分别在1 238 cm-1、1 232 cm-1处酰胺Ⅲ带(N-H弯曲振动)和1 450 cm-1附近的CH3反对称弯曲振动[12].

图2 FS和PS的紫外光谱图

图3 PS和FS的红外吸收光谱图
其中酰胺Ⅰ带、酰胺Ⅱ带和酰胺Ⅲ带与胶原的三股螺旋结构密切相关,Albu等[13]提出了两种检测胶原的三股螺旋结构完整性的方法.第一种方法是计算酰胺Ⅲ带和1 450 cm-1处的吸光度的比值即AⅢ/A1450,当比值约为1.00时表明结构保留,反之,比值远小于1.00时则说明胶原已变性,如明胶的比值为0.59[14];第二种方法是计算酰胺Ⅰ和Ⅱ带所在波数的差值即Δv=vⅠ-vⅡ,当差值远大于100 cm-1时表示胶原已变性.根据上述方法对PS的三股螺旋结构的完整性进行评估.PS的Δv值和AⅢ/A1450值分别为106 cm-1和1.008 6,类似于FS(99 cm-1、1.008 8),说明浸酸皮胶原的三股螺旋结构完整,与鲜牛皮胶原具有相同的化学结构.
2.4 表观粘度分析
图4为FS和PS溶液的表观粘度与剪切速率的关系曲线.当剪切速率在0~0.2 s-1的范围内粘度急速下降,0.2~1.0 s-1的范围内粘度逐渐趋于平缓,所提取胶原溶液整体均呈现出剪切变稀行为.由于胶原分子在溶液中主要以稳定的聚集形式存在,当受到剪切作用时,首先胶原聚集体间的缠结逐渐被破环,胶原聚集体解体;随后胶原分子间物理缠结被破坏,使得胶原易于滑动,导致剪切粘度明显降低,最终胶原聚集体完全解离为胶原分子且胶原分子沿剪切力方向发生取向性,因此在高剪切速率范围内剪切粘度趋于常数[15].此外,在0.1 s-1剪切速率时,FS溶液的表观粘度(0.114 Pa·s)略大于PS溶液的表观粘度(0.109 Pa·s).因此,FS聚集体缠结较多,结构较为紧密.

图4 PS和FS溶液的表观粘度曲线
2.5 粘度测试数学模型拟合分析
图5为Ostwald-de Waele、Carreau和Cross模型对FS和PS溶液的表观粘度数据的拟合曲线.由图5可知,三种模型的拟合曲线和实验曲线均具有良好的叠加,说明了三种模型都能较好地描述PS和FS溶液的表观粘度曲线.表1是三种模型拟合所得到的参数.比较三种模型的重合性和回归系数R2,较好的重合性、较高的R2值和较低的标准差SD值说明Cross模型可以更为准确地描述胶原溶液的稳态剪切流动.
从表1可知,非牛顿指数n、m和a均小于1,表明两种胶原溶液为典型的假塑性流体,均具有剪切变稀的特点.Ostwald-de Waele模型拟合所得粘度相关系数K值以及Carreau和Cross模型拟合所得零剪切粘度η0和η0′,表明PS溶液的粘度大于FS溶液的粘度,与实验结果一致,证实了浸酸皮胶原聚集体缠结较多,结构较为紧密.此外,由Carreau和Cross模型拟合所得无穷剪切粘度η∞和η∞′接近于0可知当剪切速率无穷大时,胶原分子沿剪切力方向发生取向性,分子间摩擦力非常小,因此粘度趋于0.

(a)PS溶液的表观粘度数据拟合曲线
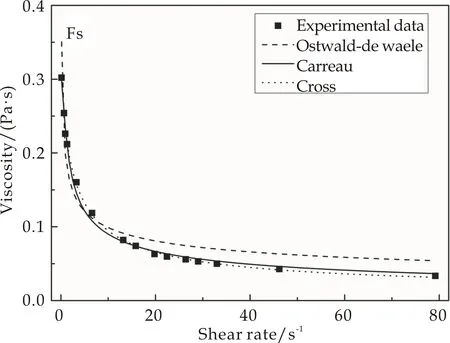
(b)FS溶液的表观粘度数据拟合曲线图5 Ostwald-de Waele、Carreau和Cross模 型对PS和FS溶液的表观粘度数据拟合曲线表1 Ostwald-de Waele、Carreau和Cross模型拟合所得到的参数

ModelParameterSamplePSFSOstwald-de WaelemodelKnR2SD0.205±0.0070.712±0.0190.9450.1800.194±0.0080.708±0.0250.9130.181Carreau modelη0η∞λmR2SD0.335±0.0044.807E-282.208±0.1250.609±0.0060.9990.0920.301±0.0071E-321.581±0.1800.563±0.0160.9940.092Cross modelη0′η∞′kaR2SD0.377±0.0040.026±0.0070.666±0.0550.742±0.0530.9980.0920.330±0.0080.010±0.0050.465±0.0310.777±0.0410.9990.081
2.6 胶原热动态分析
图6是PS和FS溶液的热容与温度的关系曲线.典型的胶原溶液的US-DSC曲线有两个吸热峰,分别代表胶原溶液的预转变和变性转变过程[16],相应的温度由Tm1和Tm2表示.结合图6和表2可知,PS的Tm1和Tm2均低于FS,而焓变(ΔH)表示破坏维持胶原三股螺旋结构的氢键所需要的能量,两个现象均表明浸酸皮胶原溶液的热动态稳定性略低于鲜牛皮胶原.

图6 PS和FS溶液的热容与温度的关系曲线表2 PS和FS溶液的热转变温度和焓变

SampleTm1/℃Tm2/℃ΔH/(kJ/mol)PS35.541.71.52×104 FS36.042.61.99×104
胶原的热变性过程主要是指胶原三股螺旋结构的崩塌、肽链解螺旋转变为无规卷曲结构的过程,而其中氢键作用对其热稳定性起着关键性的作用.据报道,胶原分子X位上的酰胺基与甘氨酸上的羰基之间所形成的氢键需要水分子作为架桥,而这些通过水分子作为架桥形成的氢键对胶原的稳定性起到重要的作用且有助于稳定胶原分子中缺乏脯氨酸的三股螺旋区域.胶原分子间缠结较多会导致胶原形成较大的聚集体,胶原分子间疏水作用增强,因此胶原分子内以水分子为架桥形成的氢键减少,从而胶原分子的稳定性下降,即胶原溶液中分子缠结较多,热稳定性较低.此外,胶原分子的热变性过程是一个吸热过程,受到热量传递速率的影响.胶原分子间缠结增多意味着胶原分子间间距较小,热传递速率较快会加速胶原的变性[17].通过上述分析可知,由于PS溶液中胶原聚集体缠结较多,结构较为紧密,因此以水分子为架桥形成的氢键减少且分子间热传递速率较快导致胶原溶液的热稳定性较低,表现为转变温度和焓变较小.
2.7 成纤维性能
图7是PS和FS的浊度曲线.在37 ℃下,胶原溶液的吸光度在15 min内快速增大,之后即可达到平稳的平台,说明两种胶原均具有良好的成纤维性能且成纤维速率较快.比较PS和FS的吸光度增加值可知,PS的成纤维能力较弱[18].

图7 PS和FS溶液的浊度曲线
为了更加直观地考察胶原纤维的结构,采用原子力显微镜观测胶原纤维形貌并分析胶原纤维的平均直径和直径分布,如图8所示.

(a)FS纤维的AFM图 (b)PS纤维的AFM图
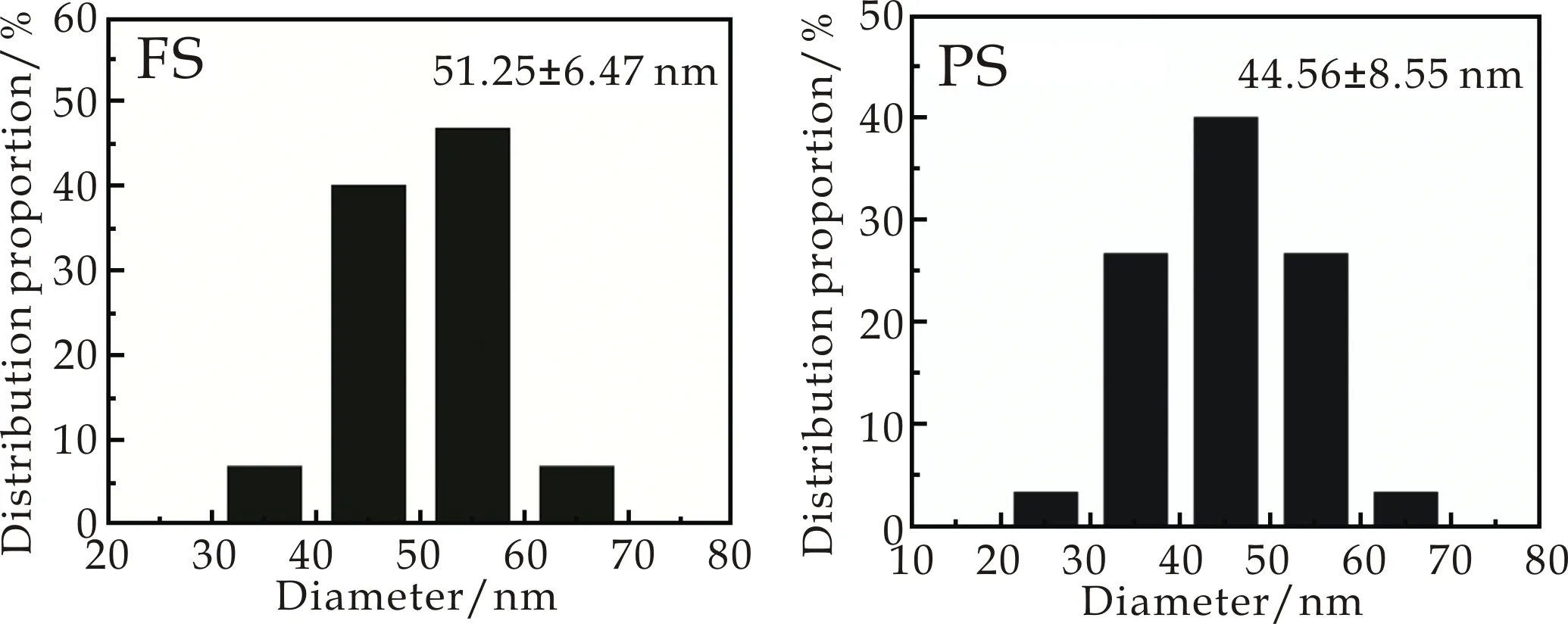
(c)FS纤维的直径分布图 (d)PS纤维的直径分布图图8 PS和FS纤维的AFM图 和纤维直径分布图
由图8(a)、(b)可看出,FS和PS胶原纤维均呈现出成熟的纤维立体网状结构,胶原纤维互相紧密缠绕在一起且排列随机杂乱,无方向性.对比FS和PS胶原纤维的直径,可知PS胶原纤维平均直径为44.56±8.55 nm,略细于FS胶原纤维(51.25±6.47 nm),因此表现为吸光度较低.其中,PS胶原纤维主要集中分布在30~60 nm,而FS胶原纤维主要集中分布于40~60 nm.
3 结论
以制革过程中间物——浸酸牛皮为原料提取胶原,并与鲜牛皮所提取的胶原进行对比.通过电泳、红外、紫外结果可知两种胶原结构基本一致,具有完整的三股螺旋结构且纯度较高.旋转粘度计测定结果表明,浸酸皮胶原溶液中聚集体缠结较多因此热稳定性较鲜牛皮胶原溶液偏低、粘度比鲜牛皮胶原溶液稍高;此外二者体系均属于假塑性流体、呈现剪切变稀行为且成纤维能力相近,说明浸酸过程对胶原的三股螺旋结构无明显影响;浸酸牛皮作为提取胶原的原材料是可行的且操作简单、周期较短.
