LRBA基因突变2例病例报告
2019-03-28刘璐瑶董小龙孙金峤王晓川
刘璐瑶 王 莹 董小龙 林 丽 孙金峤 王晓川
1 病例资料
1.1 病例报告
例1 男,13岁,因“PLT、Hb反复下降6年余”于2016年1月25日至复旦大学附属儿科医院(我院)就诊。
患儿于2009年无明显诱因出现皮肤淤青,就诊于当地医院,发现PLT、Hb下降,考虑自身免疫性贫血、PLT减少性紫癜。曾给予激素、免疫球蛋白治疗后PLT升至正常,溶血性贫血较前好转,激素减量时病情易反复。2010年7月在上海市某儿童医院行骨髓细胞学检查:骨髓增生明显活跃,粒红比倒置,粒系增生相对减低,红系增生明显活跃,巨核系增生活跃。2013年就诊于我院,考虑为自身免疫性溶血性贫血,免疫功能检查示免疫球蛋白减低。2013至2016年接受激素治疗,曾患真菌性肺炎。
患儿系G1P1,足月剖宫产。父母身体健康,否认近亲结婚及家族相关遗传性疾病史。既往有丙种球蛋白、红细胞悬液、冷沉淀、白蛋白和纤维蛋白原输注史。
体格检查:神志清楚,反应良好,精神可,贫血貌。两肺呼吸音粗,未及啰音。腹壁松弛,腹部皮纹明显。肝肋下3 cm,剑突下3 cm,质韧,有触痛。脾肋下6 cm,质韧,有触痛。腹部叩诊鼓音,腹壁静脉显露,移动性浊音阴性。因长期服用激素表现为库欣貌,向心性肥胖,发育落后。
实验室检查:血常规WBC 7.3×109·L-1,L 1.8×109·L-1,N%为70.9%,CRP 16 mg·L-1;RBC 2.42×1012·L-1,Hb 55 g·L-1,网织红细胞比例14.3%,抗C3阳性,抗IgG阳性,Coombs试验血型单特异性抗体阳性,直接抗人球蛋白试验阳性提示溶血性贫血。血生化ALT 206 IU·L-1,AST 255 IU·L-1;自身抗体阴性。外周血白细胞中EBV-DNA阳性(4.13×104拷贝数·mL-1);真菌G试验89.0 pg·mL-1;肺泡灌洗液宏基因检测真菌弱阳性。
例2 男,3个月2 d,因“反复PLT减少3个月”于2018年8月7日至我院就诊。
患儿出生后因“气促,吐沫10 min”于外院就诊。X线胸片提示新生儿肺炎,血常规PLT 19×109·L-1, CRP 9 mg·L-1, WBC 28.2×109·L-1,N% 85.4%,予抗感染,静脉输注丙种球蛋白2.5 g、PLT 60 mL,泼尼松1 mg·kg-1口服。黄疸明显,诊断为新生儿高胆红素血症。病程中无明显诱因出现发热,伴外耳道疖肿破溃流脓,全身皮肤可见散在出血点。骨髓穿刺结果提示:骨髓增生活跃(粒系、红系),PLT较少见,可见巨核细胞,嗜酸性粒细胞比值偏高。生后有反复腹泻,蛋花样便,1月龄停母乳后好转。
患儿系G2P1,41+1周产钳助产出生,出生体重3 850 g,出生无窒息抢救史。母亲第1胎于孕10周胎停。父母身体健康,否认近亲结婚及家族相关遗传性疾病史。
体格检查:神志清楚,反应良好,精神可,发育正常;全身皮肤可见散在少许针尖大小出血点,全身浅表淋巴结未触及肿大,腹软,肝脾肋下未触及;四肢肌力、肌张力可,中枢神经系统检查未见异常。
实验室检查:血常规WBC 9.6×109·L-1,L 6.1×109·L-1,N 1.02×109·L-1,嗜酸性粒细胞1 860个/μL,Hb 94 g·L-1,RBC 3.98×1012·L-1,PLT 10×109·L-1,网织红细胞百分比 1.8%,CRP<8 mg·L-1。Coombs试验阴性。尿有机酸质谱结果显示,3-羟基-苯乙酸-2、4-羟基-苯乳酸-2水平略高于正常。静脉血EBV DNA阴性,CMV DNA阴性;外周血白细胞EBV DNA阴性。自身抗体阴性。
1.2 影像学检查 例1 CT报告显示两肺广泛多发斑片、结节影,纵隔多发肿大淋巴结影(图1A),考虑真菌感染的可能。B超报告胆囊内密集占位,提示真菌感染(图2A);肝肿大,肝脏质地欠佳,巨脾(图2B),脾静脉增宽。例2 B超报告肝质地欠佳(图2C);双侧颈部探及淋巴结,双侧腋下淋巴结肿大;双侧腹股沟淋巴结轻度肿大;脾脏未见明显局灶性占位。腹部肠腔内大量气体。

图1例1CT检查结果
注 A:两肺广泛多发斑片、结节影,纵隔多发肿大淋巴结影。B:两肺透亮度尚可,广泛多发斑片、结节影较前有明显好转
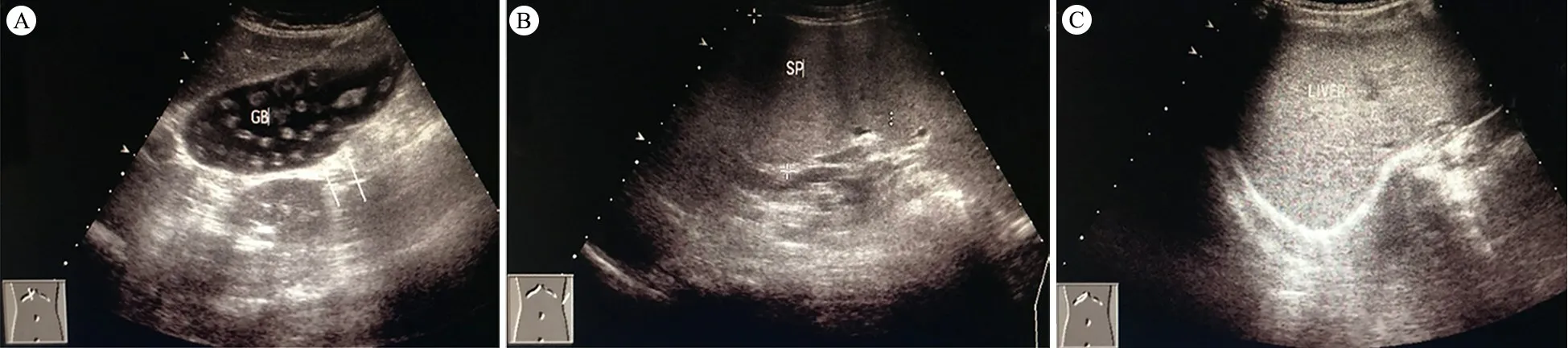
图22例患儿B超检查结果
注 A:例1 胆囊内密集占位;B:例1巨脾;C:例2肝质地欠佳
1.3 免疫功能评价
1.3.1 常规免疫检查 采用免疫比浊法检测免疫球蛋白IgG、IgM和IgA的水平。使用荧光标记单克隆抗体分别标记外周血淋巴细胞亚群,其中抗CD19标记B细胞,抗CD3、抗CD8抗体标记CD8+T细胞,抗CD3、抗CD4抗体标记CD4+T细胞,抗CD16CD56抗体标记NK细胞,用流式细胞仪(BD Biosciences, San Jose, CA)分析结果。
1.3.2 淋巴细胞精细分型 取混匀的 50 μL肝素抗凝全血于流式管中,加入不同荧光素标记的小鼠抗人APC-CD19、BV421-CD27、BV510-IgD、PE-CD24和PERCP-CY5.5-CD38检测B淋巴细胞不同分化发育亚群;使用小鼠抗人PE-TCRαβ、BV421-TCRγδ 、PERCP-CY5.5-CD3、FITC-CD4、BV510-CD8、APC-CD27和PE-CY7-CD45RA检测T淋巴细胞不同分化发育亚群;混匀后室温避光孵育;加入裂红素裂解红细胞。使用PBS缓冲液重悬后用BD Diva 软件上机获取数据并分析。
1.3.3 免疫功能评价 例1 IgG、IgA和IgM均下降,CD3+T淋巴细胞和CD8+T淋巴细胞增多,CD4+T淋巴细胞、B淋巴细胞和NK细胞减少(表1)。例2 IgG升高,IgA和IgM在正常范围,CD4+T淋巴细胞增多,CD8+T淋巴细胞减少,B淋巴细胞和NK细胞均在正常范围(表1)。精细分型结果提示2例初始B细胞比例增高、记忆性B细胞降低(图3、表1);例1浆细胞(0.1%)、初始CD4+T细胞(1.6%)和CD8+T细胞(6.9%)比例下降,中央记忆性CD4+T细胞(81.5%)和CD8+T细胞(37.2%)比例增高(图3、4);例2 T淋巴细胞亚群精细分型大致正常(图4)。
1.4 基因检测 取得患儿父母知情同意后,抽取患儿及其父母外周静脉血2 mL(EDTA 抗凝),提取基因组 DNA(QIAGEN公司),采用高通量测序法行全外显子组测序。采用Sanger法和荧光定量PCR分别对点突变和缺失突变进行验证。2例均为LRBA基因复合杂合突变(图5A)。例1为c.1933C>T(p.R645X,母源)杂合突变和29号外显子缺失杂合突变(父源),c.1933C>T导致645位编码精氨酸的密码子突变为终止密码子,导致蛋白翻译提前终止,引起LRBA蛋白的异常截短,该突变位点在HGMD数据库中尚未报道(图5)。例2为c.3778G>C(p.A1260P, 母源)和c.1570G>A(p.G524S, 父源)复合杂合突变(外院检测)。c.3778G>C(p.A1260P)在HGMD数据库中尚未报道,经mutation taster预测为致病突变;c.1570G>A(p.G524S)为HGMD数据库中已报道的致病突变(CM1720278)。
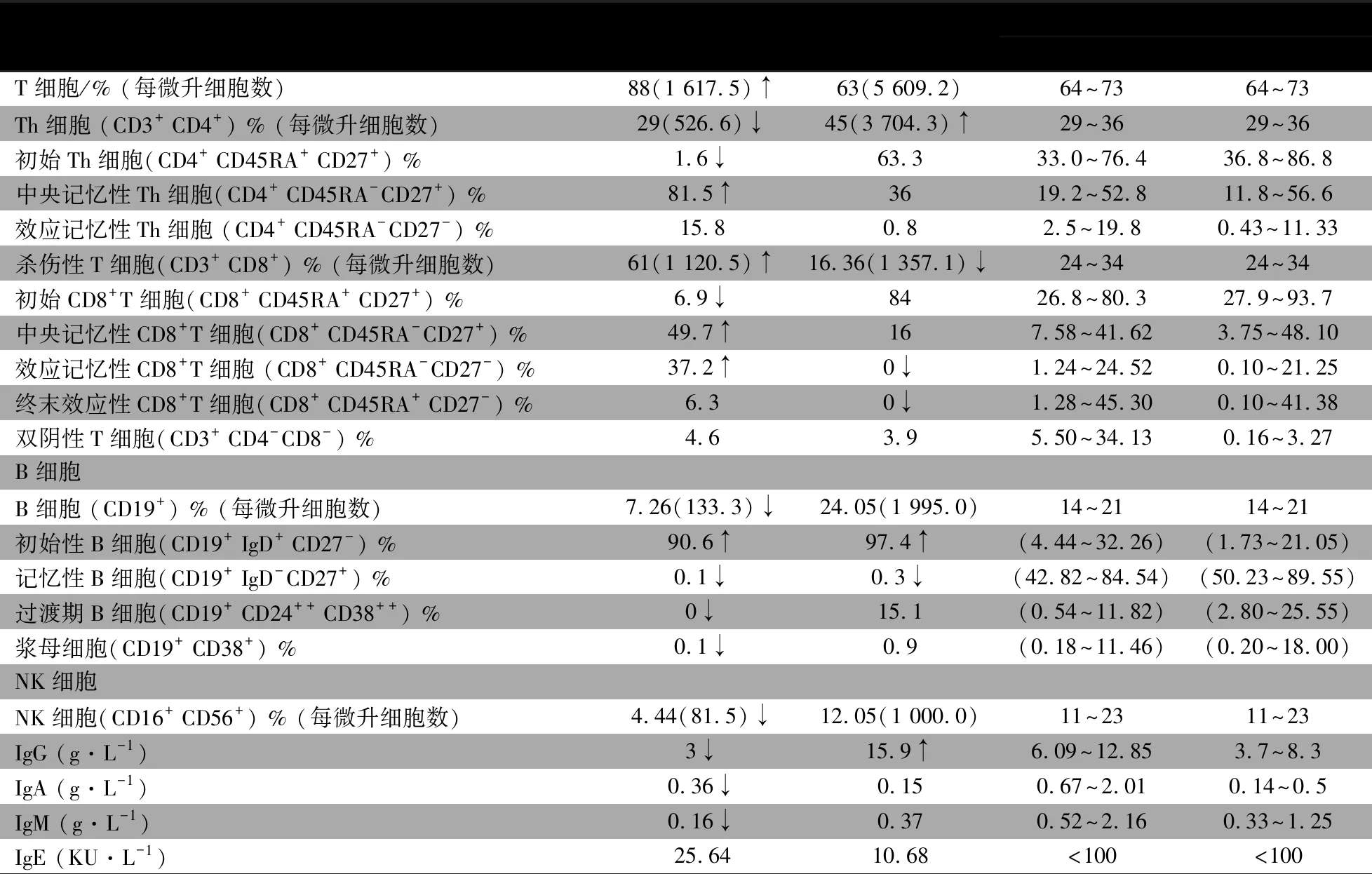
表1 2例患儿免疫功能检查
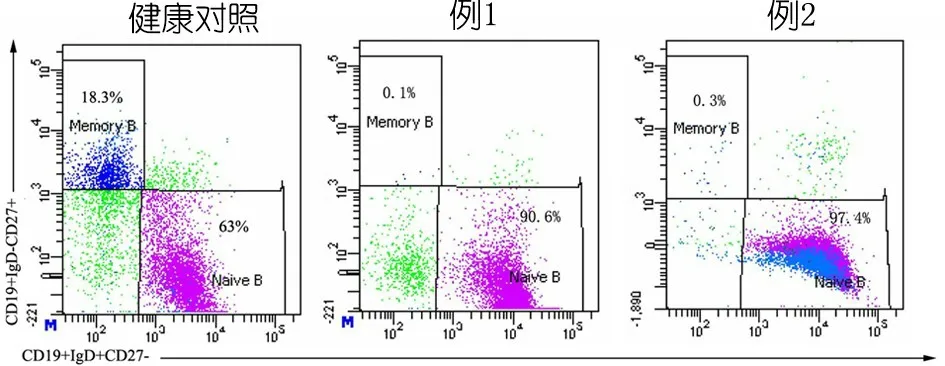
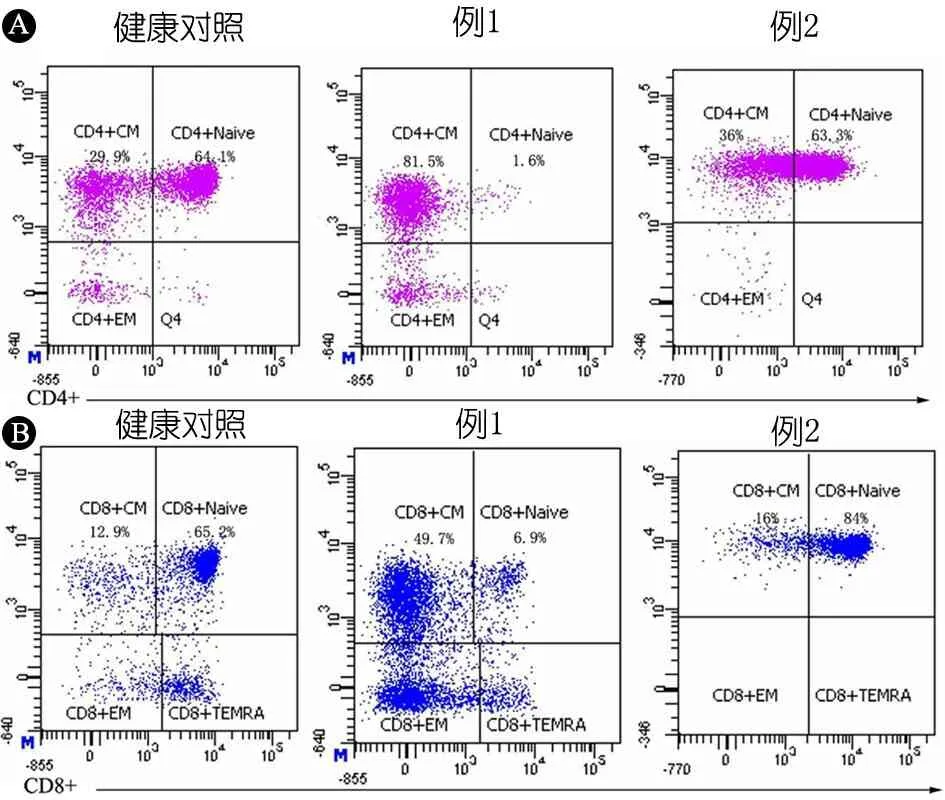
图3 2例患儿B淋巴细胞亚群精细分型结果
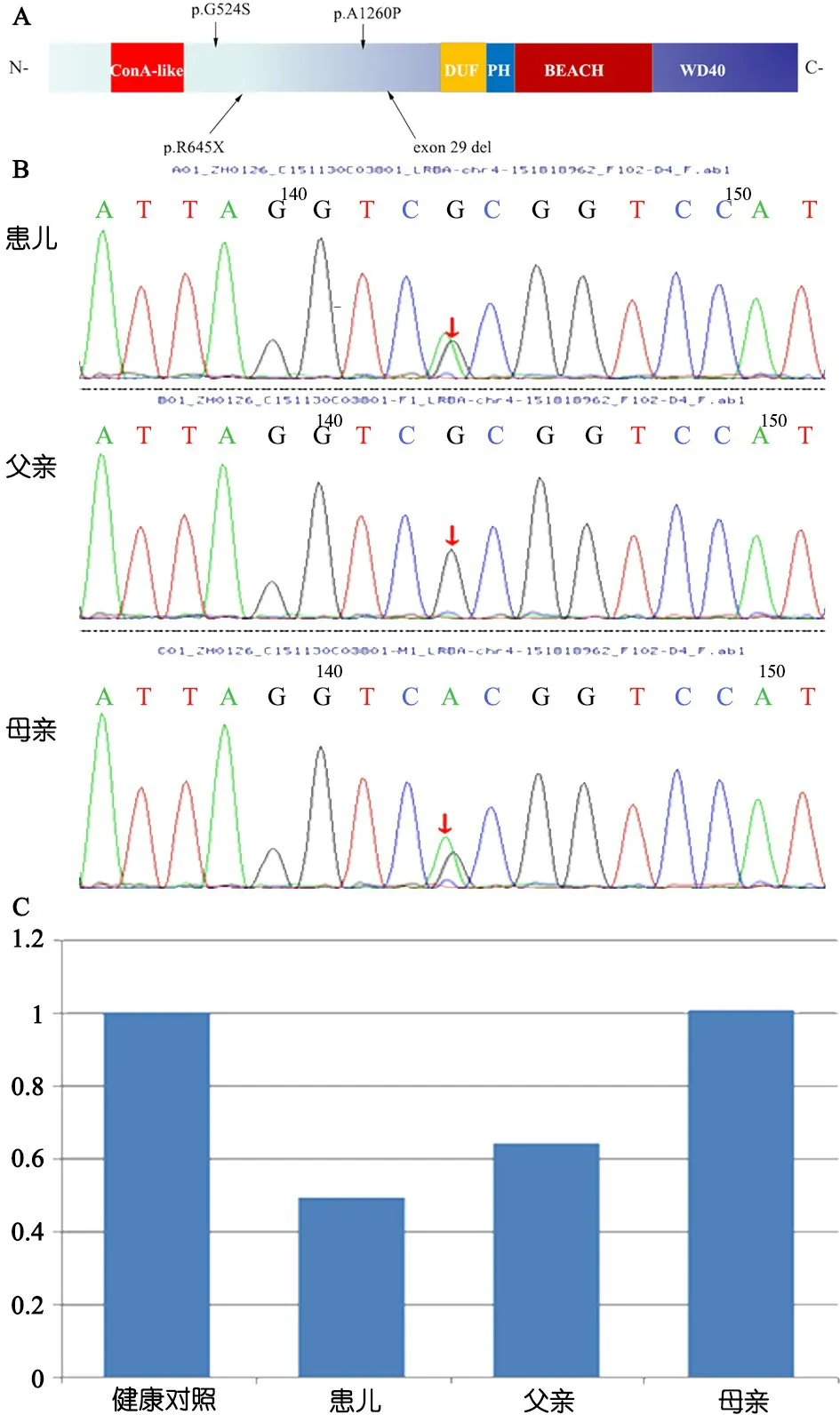
图4 2例患儿CD4+ T和CD8+ T淋巴细胞亚群精细分型结果
图5基因检测结果
注 A:LRBA蛋白结构示意图及2例患儿突变分布(例2:p.G524S和 p.A1260P);B:例1及其母亲均检测到LRBA基因15号外显子c.1933C>T(p.R645X)杂合突变;C:例1家系LRBA(exon 29)荧光定量检测结果,例1及其父亲均检测到LRBA基因 29号外显子杂合缺失
1.5 治疗 例1入院后完善相关检查,给予甲泼尼龙(80 mg q12 h)、免疫球蛋白(共40 g)抑制免疫反应、减少红细胞破坏;输注红细胞;头孢哌酮舒巴坦、伏立康唑抗感染,效果不佳,后加用卡泊芬净抗真菌治疗,肺部真菌感染症状好转(图1B),Hb及PLT逐渐恢复正常,网织红细胞逐渐下降,患儿病情好转,定期输注丙种球蛋白。因家庭原因未进行造血干细胞移植(HSCT)。
例2予静脉输注丙种球蛋白2.5 g及PLT,PLT较前无明显上升。目前已行HSCT,临床好转中。
2 讨论
LRBA (LPS-responsive beige-like anchor protein) 缺陷是LRBA基因发生双等位基因功能缺失性突变所引起的一种原发性免疫缺陷病[1]。LRBA基因位于4q31.3,编码LRBA蛋白,是BEACH-WD40蛋白家族成员之一,普遍存在于人体内,在免疫细胞尤其是淋巴细胞中高度表达。目前LRBA缺陷具体的发病机制仍然不清楚,但是相关研究发现LRBA蛋白与蛋白激酶A结合,主要调节胞内囊泡的转运过程,有助于免疫效应分子如细胞毒性T淋巴细胞抗原4(CTLA-4)的分泌和膜沉积过程[2]。CTLA-4在活化的T细胞和调节性T细胞(Tregs)上表达,通过与共刺激分子CD28(CD80和CD86的配体)竞争或者直接通过跨细胞胞吞作用去除抗原递呈细胞上的这类配体,从而发挥免疫抑制作用[3, 4]。 LRBA蛋白能够稳定CTLA-4胞浆内尾端,防止其被溶酶体降解,LRBA缺陷导致活化的CD4+T细胞和Tregs上CTLA-4 的水平降低,从而出现一系列免疫失调的表现[2]。
LRBA缺陷在2012年[1]被提出后最先被归为常见变异性免疫缺陷病(CVID)[1, 5]。目前多个研究发现部分LRBA缺陷的患者表现为自身免疫性疾病,如炎症性肠病、类免疫失调-多发内分泌病-肠病-X染色体连锁(IPEX)综合征和类自身免疫性淋巴增生综合征(ALPS)等[6, 7]。LRBA缺陷主要表现为一种表型广泛的免疫综合征,在2017年免疫缺陷病分类中将其归为免疫失调性疾病[8]。LRBA缺陷的临床表现复杂多样,主要包括低丙种球蛋白血症、自身免疫性疾病、反复感染、免疫失调、肝脾肿大、肺部疾病以及易感EBV等,其中低丙种球蛋白血症和慢性腹泻最常见[9, 10]。LRBA缺陷患者免疫功能的异常主要包括IgG抗体产生减少,特异性抗体免疫应答缺陷,T淋巴细胞的增殖和活化功能缺陷,Treg细胞减少以及B淋巴细胞的自噬减少等。大多数LRBA缺陷的患者B淋巴细胞亚群的计数减少,尤其是类别转换记忆性B细胞和浆细胞[1, 11]。
本文报告的2例LRBA缺陷患儿均有反复感染、血细胞减少和腹泻等自身免疫性疾病的表现。淋巴细胞亚群精细分型结果提示,2例患儿初始B细胞比例明显增高,记忆性B细胞比例降低。例1还表现为免疫球蛋白IgG、IgA、IgM水平下降,CD3+T和CD8+T淋巴细胞增多,CD4+T淋巴细胞、B淋巴细胞和NK细胞减少,浆细胞、初始CD4+T和CD8+T淋巴细胞比例下降,中央记忆性CD4+T和CD8+T淋巴细胞比例明显增高等。例1的临床表现以及免疫表型符合之前所报道的LRBA缺陷患儿的表现,但是例2与例1的表型不完全一致,这种现象说明LRBA缺陷复杂多样的临床表现可能受到发病的年龄、感染以及外界环境因素等的影响。儿童若出现早发型低丙种球蛋白血症、严重的自身免疫性疾病、炎症性肠病、淋巴组织增生和反复呼吸道感染等临床表现,需考虑LRBA缺陷可能。
目前LRBA缺陷的治疗手段主要包括阿巴西普靶向治疗和HSCT。其中,HSCT也可以用于伴发严重疾病的患者[12]。阿巴西普是一种 CTLA-4免疫球蛋白融合药物,通过与抗原提呈细胞上的 CD 80/86 结合抑制T 细胞激活,目前用于治疗类风湿关节炎。之前有报道1 例CTLA-4 功能性突变的女孩在接受阿巴西普治疗后,其自身免疫症状有所改善,FOXP3表达升高,Treg细胞功能恢复正常[13]。部分LRBA缺陷的患者接受阿巴西普治疗后,间质性肺病、自身免疫性疾病以及免疫表型都有了显著的改善[14]。但是血细胞减少等临床表现对阿巴西普治疗的反应性比较差。
HSCT是根治LRBA缺陷的主要手段,相关临床研究[12, 15-18]报道,已有12例LRBA缺陷患者接受了HSCT治疗,总生存率为67% (8/12), 其中有6例临床表现好转[19]。本文例2行HSCT,目前正处于临床好转阶段,例1因家庭原因目前还没有接受HSCT治疗。目前接受HSCT治疗的LRBA缺陷的病例仍有限,效果尚待进一步研究。对于部分临床表现复杂、相关并发症严重的LRBA缺陷的患儿,若常规治疗无效,可尽早行HSCT。
